Nivel de manejo del médico general: Diagnóstico: sospecha. Tratamiento: inicial. Seguimiento: derivar.
Aspectos esenciales
-
Condición de cronicidad de crisis epilépticas paroxísticas (2 o + de similar presentación).
-
Causas: idiopatica o secundaria.
-
Evaluar patrón de presentación y clasificación en simple o compleja, focal o de rápida generalización.
-
Diagnóstico es clínico asociado a la presencia ideal de un testigo. Exámenes son complementarios.
-
Descartar causas secundarias de urgencia, otras causas de alteración de conciencia o convulsiones y derivar.
Caso clínico tipo
Hombre de 45 años, es traído al servicio de urgencias tras haber sido encontrado en el suelo con desviación de cabeza y movimientos repetitivos de extremidad superior derecha hacia la derecha, con posterior presentación de movimientos descritos como sacudidas con pérdida de conciencia posterior. Se desconoce la duración del episodio. Paciente refiere posterior al cuadro se encontraba confuso y somnoliento. Al examen físico se evidencia cianosis leve y lesiones de mordedura de lengua en la zona lateral.
Definición
Corresponde a la recurrencia de crisis epilépticas paroxísticas sin tener un desencadenante identificable de inmediato, causadas por la descarga sincrónica y exagerada de un grupo de neuronas que puede progresar o no a la descarga sincrónica de ambos hemisferios.

Etiología
Causas más frecuentes por edad:
<20 años: asfixia perinatal, infección SNC, trastornos metabólicos, traumas, malformaciones arteriales, criptogénica, idiopáticas.
20-45 años: TEC, malformación arterio-venosa, infecciones SNC (cisticercosis, abscesos cerebrales), abuso de sustancias, abstinencia de alcohol, idiopáticas.
>45 años: tumores, enfermedad cerebro-vascular (ECV), cisticercosis, enfermedades neurodegenerativas.
CAUSAS PRINCIPALES DE LAS EPILEPSIAS
|
A. HEREDITARIAS |
Epilepsias genéticamente determinadas. |
|
B. CONGÉNITAS (Hereditarias o Adquiridas) |
Displasias o disgenesias cerebrales Algunos tumores cerebrales Lesiones intraútero Malformaciones vasculares Sindromes neurocutáneos (neurofibromatosis, enfermedad de Sturge-Weber, esclerosis tuberosa) Anomalías cromosómicas (síndrome de Down-trisomía 21, cromosoma 20 en anillo, síndrome de Angelman-deleciones en cromosoma 15, trisomía o microdeleciones en otros cromosomas) Trastornos congénitos del metabolismo (aminoacidurias, leucodistrofias) Miopatías congénitas tipo Fukuyama Epilepsias mioclónicas progresivas |
|
C. ADQUIRIDAS |
Traumatismos Lesiones posquirúrgicas Lesiones postinfecciosas Infarto y hemorragias cerebrales Tumores Esclerosis del hipocampo (del lóbulo temporal) Tóxicos (alcohol y otras drogas) Enfermedades degenerativas (enfermedad de Alzheimer y otras demencias) Enfermedades metabólicas adquiridas |
Compendio de Medicina Interna. C. Rozman. V edición. Editorial Elsevier.
Epidemiología
-
17 por 1.000 hab. en Chile, con una incidencia de 114 por 100.000 hab. al año
-
Frecuencia mayor en <20 y >60 años
-
Mortalidad 2 a 3 veces la general.
Fisiopatología
Las crisis son producidas por un desequilibrio excitatorio/inhibitorio del SNC, que deriva en hiperexcitabilidad celular, mediado por aumento de entrada de Ca2+ y Na+ a la célula, activación de NMDA, y disminución del efecto GABA en un grupo focalizado de neuronas (crisis focal). Ésta puede propagarse a través de la excitación de neuronas contiguas por el desbalance de Neurotransmisores a otras áreas del cerebro, pudiendo tener compromiso de un hemisferio y afectar en forma bilateral (crisis de rápida progresión o generalizada). La clínica se producirá dependiendo de la zona inicialmente excitada a nivel focal, evolucionando posteriormente a compromiso de conciencia si llega a afectar ambos hemisferios.
Diagnóstico
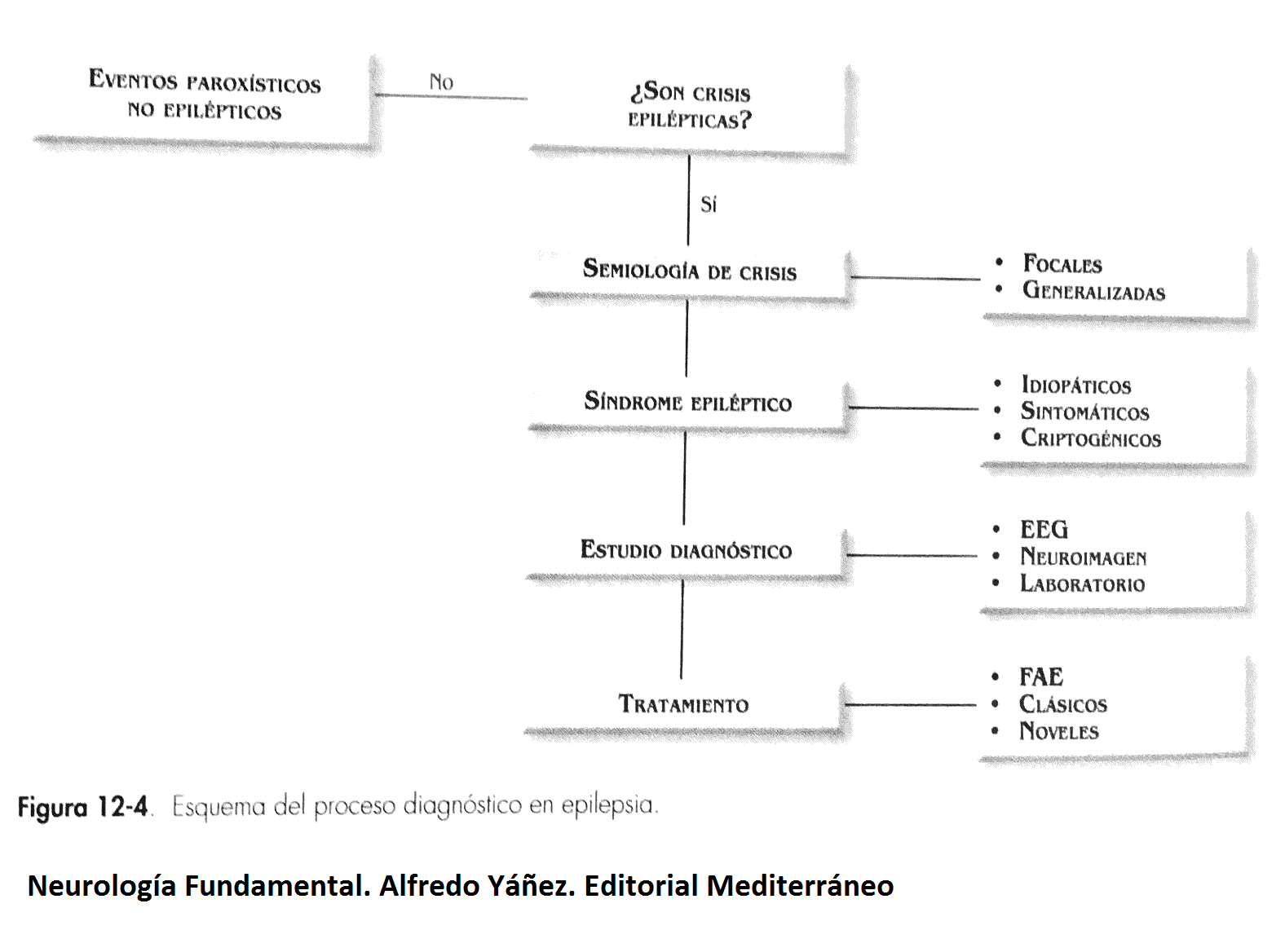
La sospecha es eminentemente clínica. El elemento esencial para la sospecha es la presencia de un testigo. Se debe evaluar eventos desencadenantes asociados al episodio (consumo de medicamentos, alcohol, drogas, antecedente de trauma, cefaleas recurrente, etc). Se debe caracterizar el episodio y lograr al menos una clasificación del episodio de crisis, destacando el estereotipo de crisis en relación a cuadros similares anteriores. El diagnóstico definitivo lo hace el especialista.
Se pueden clasificar las crisis inicialmente en:
- Focales simples (o anterior parcial simple): se mantiene nivel de conciencia. Signo o síntoma característico del lóbulo comprometido. Pueden ser motoras (aumento de tono, movimientos repetitivos, automatismo), sensoriales (sonidos, cacosmia, escotomas o visión de halos), psíquicos (deja vú, miedo, despersonalización), con o sin síntomas autonómicos (piloerección, sudoración). Pueden progresar a generalizadas. Puede darse la llamada progresión Jacksoniana (compromiso inicial a nivel de extremidades inferiores con progresión hacia extremidades superiores o viceversa) con posterior parálisis o paresia de las extremidades comprometidas (Parálisis de Todd).
- Focal compleja (o anterior parcial compleja): crisis focal que se acompaña de algún nivel de alteración de conciencia. Puede presentar automatismos (ej. chupeteo), con confusión posterior. No confundir con crisis de ausencia.
- De rápida progresión o generalizada: Se afectan simultáneamente a ambos hemisferios cerebrales desde un inicio. Tiene compromiso de conciencia. Corresponden a las anteriormente llamadas petit mal (crisis de ausencia) o gran mal (tónica-clónica). Puede ser tónico-clónica, tónico, clónica, ausencias o atonías. Suelen tener un periodo de corta duración, menor a 5 minutos (más de 5 minutos corresponde a un status epiléptico), con confusión posterior.
- Mioclónica Juvenil: Se inicia con sacudidas de extremidades, generalmente a nivel de extremidades superiores, con movimientos repetitivos, cíclicos. Pueden asociarse a eventos tónico-clónicos o de tipo ausencia. Generalmente tienen como gatillante la abstinencia de sueño o el consumo de alcohol.
Tratamiento
La decisión de iniciar el tratamiento farmacológico corresponde al especialista una vez confirmado el diagnóstico. La meta es la reducción del número y severidad de crisis, con menor cantidad de efectos colaterales. El fármaco a elección depende del tipo de crisis.
Para el inicio del tratamiento, se realiza en forma gradual de acuerdo a la posología correspondiente a cada medicamento, y considerar el balance entre la desaparición de crisis y efectos adversos. De no lograrse con una droga con dosis máxima tolerable, se decidirá cambiar por un segundo medicamento en forma gradual con retiro al momento de lograr los niveles terapéuticos con la segunda droga. Eventualmente de no conseguirse disminución de las crisis, evaluar combinación de medicamentos o asociación de un fármaco de segunda línea.
- Crisis focales: Carbamacepina, Fenitoína, Lamotrigina, Levetiracetam.
- Crisis Generalizadas Tónico Clónicas: Carbamazepina, Ac. Valprocio, Lamotrigina, Fenitoina, Fenobarbital.
- Crisis Mioclónias: Ac. Valproico, Levetiracetam, Topiramato.
Importante: NO UTILIZAR Carbamacepina, Oxcarbacepina, Gabapentina en crisis mioclónicas o de ausencia, ya que pueden desencadenar crisis.
Siempre evaluar estudio neuroimagenológico para descartar lesiones de tipo estructurales, que sean eventualmente susceptibles de resecar en forma quirúrgica. Considerar también en casos refractarios al uso de medicamentos con alto indice de efectos secundarios.
Seguimiento
Diagnóstico final, tratamiento y seguimiento corresponden al especialista.
Seguimiento en el caso de uso de Carbamacepina y Ac. Valproico con pruebas hepáticas y hemograma.
Bibliografía
1. Compendio de Medicina Interna. C. Rozman. V edición. Editorial Elsevier
2. Epilepsia. Neurología fundamental. A. Yañez. 2º ed. Editorial Mediterraneo
