La piel es el órgano más grande del cuerpo humano, representando aproximadamente el 6% del peso corporal. Es una estructura formada por varios tipos de tejidos que provienen del endodermo y el mesodermo embrionarios. Corresponde a una interfase entre el medio interno y externo, con múltiples funciones esenciales para la vida humana, como son la termorregulación, la función de barrera contra las infecciones y agentes físicos (radiación, calor, frío), función inmunológica, reparación de heridas, síntesis de vitamina D, excreción de sustancias a través del sudor, función sensorial, entre otras.
Composición:
La piel se compone de 3 capas (epidermis, dermis e hipodermis), incluidos los anexos cutáneos (Fig. 1).
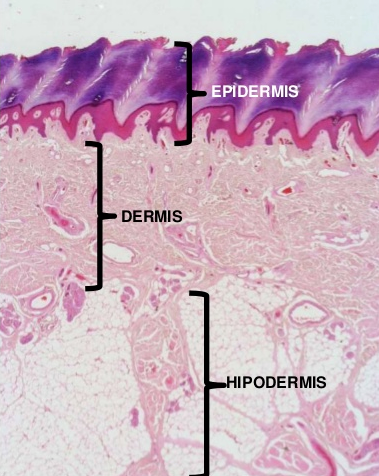
Figura 1: Las 3 capas de la piel.
A. Epidermis
Corresponde a un epitelio pluriestratificado plano y queratinizado (Fig.2). Es un tejido de alto recambio celular, con una importante diferenciación entre sus distintas capas. La célula más abundante es el queratinocito, existiendo también melanocitos, células de Langerhans y células de Merkel.
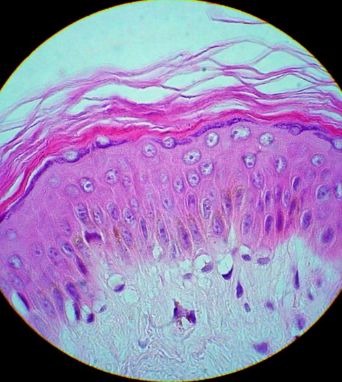
Figura 2: Epidermis.
Queratinocito
Los queratinocitos o células escamosas, se originan del ectodermo y constituyen alrededor del 80 a 90% de la epidermis. Gracias a la presencia de uniones estrechas y desmosomas entre estas células es posible mantener la estabilidad y permeabilidad selectiva del epitelio. Su función principal es la queratinización, proceso de diferenciación celular en el cual el queratinocito sufre cambios metabólicos y morfológicos con el fin de producir y acumular queratina; este proceso de diferenciación ocurre desde la base hacia la superficie, y los queratinocitos inicialmente poliédricos van perdiendo sus núcleos y adquiriendo una morfología aplanada,para quedar finalmente como grandes acúmulos de queratina en la superficie. Este proceso de diferenciación celular,en la piel sana, dura aproximadamente 28 a 30 días.
En función de estos cambios morfológicos, podemos distinguir 5 estratos en la epidermis, que de profundo a superficial son (Fig.3):
- Estrato basal o germinativo
- Estrato espinoso o mucoso de Malpighi
- Estrato granuloso
- Estrato lúcido
- Estrato córneo
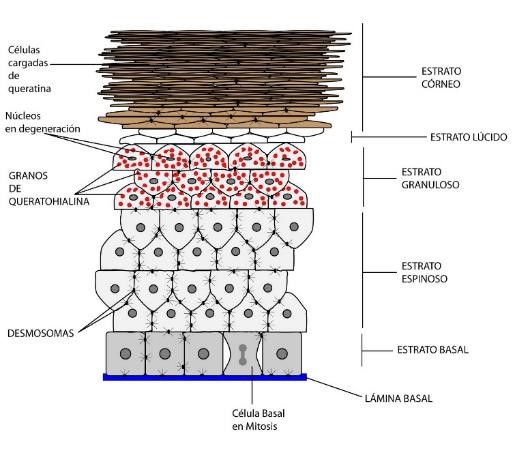
Figura 3: Esquema de la epidermis.
- Estrato basal o germinativo: formado por queratinocitos cilíndricos mitóticamente activos, responsables de la reproducción y reemplazo de las células epidérmicas (Fig.4).
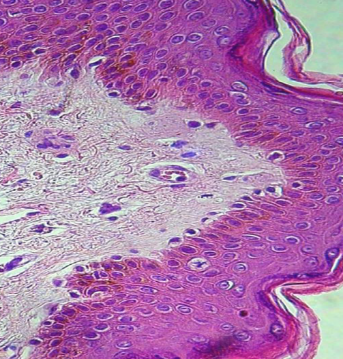
Figura 4: Estrato basal.
- Estrato espinoso o mucoso de Malpighi: formado por 5 a 10 capas de queratinocitos poliédricos, cada vez más planos hacia la superficie, unidos entre sí por desmosomas y uniones adherentes (Fig.5 ).
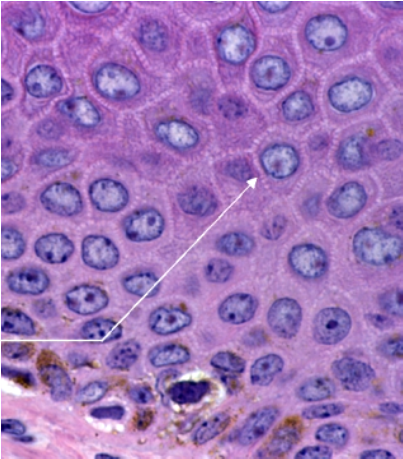
Figura 5: Estrato espinoso.
- Estrato granuloso: los queratinocitos presentan gránulos de queratohialina que contienen a los precursores para la formación de queratina, éstos gránulos son muy basófilos, lo que les da su coloración característica (Fig.6).
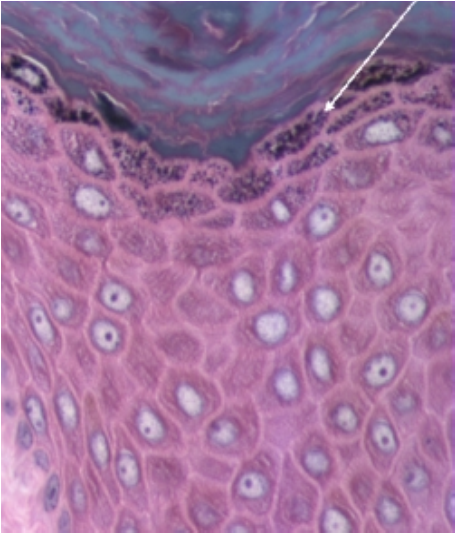
Figura 6 : Estrato granuloso.
- Estrato lúcido: sólo presente en piel gruesa, (ubicada en palmas y plantas). Está formado por eleidina, lipoproteína hidrofóbica que le confiere las características físico-químicas a este estrato (Fig. 7).
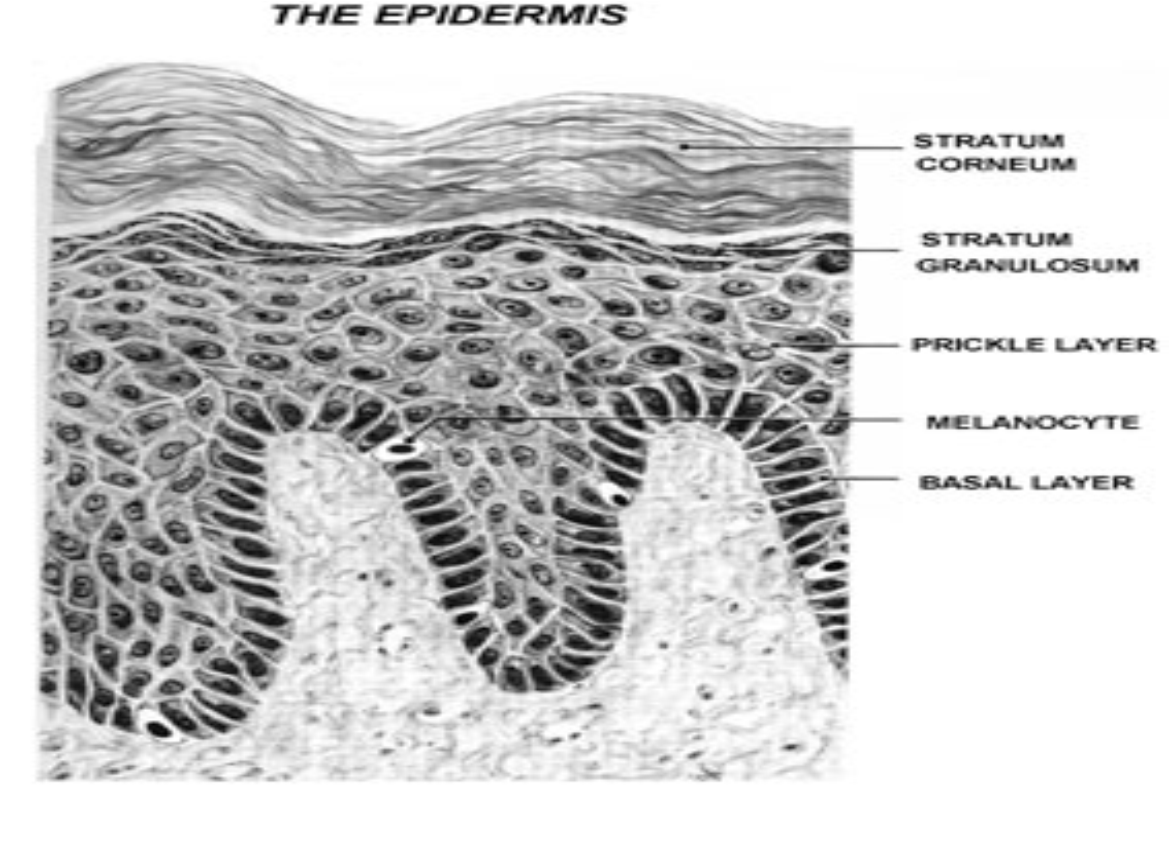 Figura 7: Estrato lúcido.
Figura 7: Estrato lúcido.
- Estrato córneo: queratinocitos aplanados sin núcleo ni organelos, sólo citoesqueleto y queratina; su grosor depende del tipo de piel, siendo mayor en las áreas de piel gruesa (Fig.8).
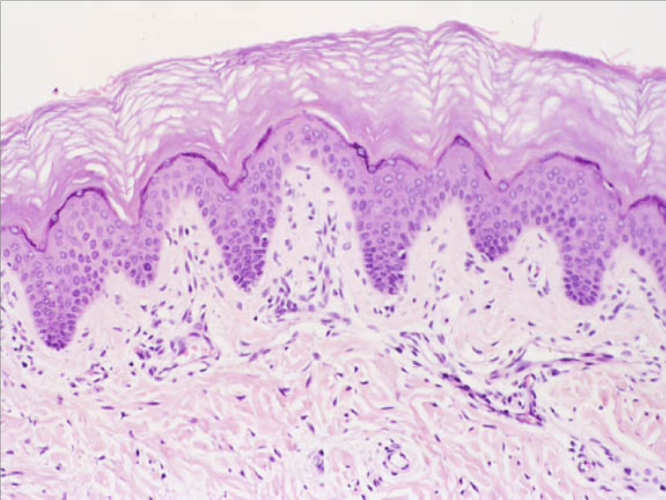
Figura 8 : Estrato córneo.
Notas:
- Mientras en la palma y las plantas hay piel gruesa, en el otro extremo está la piel delgada, que se ubica principalmente en antebrazos, párpados, labios, genitales, axilas y cuello. Esto es importante, porque por ejemplo, al usar fármacos tópicos, la penetración será mucho más intensa en estas áreas, mientras que en las de piel gruesa será baja; esto hace que se deban adecuar la potencia o concentración de los fármacos tópicos, en especial los corticoides.
- En la psoriasis (Fig.9), el ciclo celular de los queratinocitos es más rápido ,dura aproximadamente 14 días, lo que explica las alteraciones clínicas.
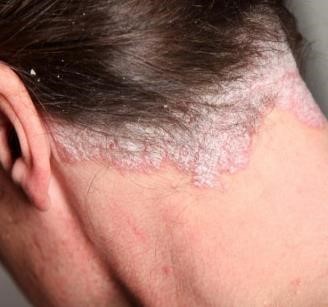
Figura 9 : Psoriasis cuero cabelludo.
Células epidérmicas no queratinocitos
Melanocitos
Célula dendrítica sintetizadora de pigmento (melanina). Deriva de la cresta neural , se encuentra principalmente en la capa basal de la epidermis (Fig.10) en una relación de hasta 1 melanocito por cada 36 queratinocitos basales y suprabasales formando la Unidad melano-epidérmica. Esta unidad está constituida por : un melanocito con los queratinocitos que reciben la melanina sintetizada por éste (Fig.11).
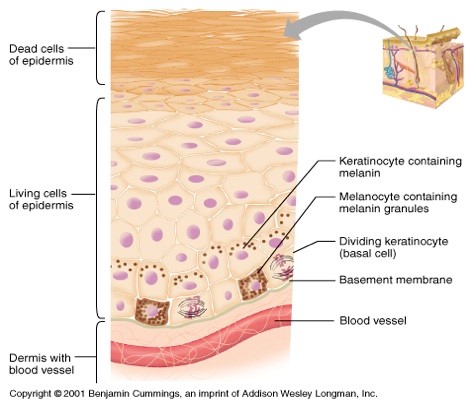
Figura 10: Ubicación de melanocitos.
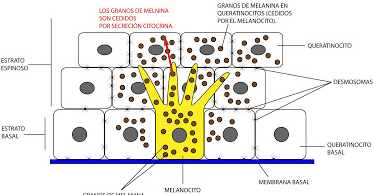
Figura 11: Unidad melanoepideérmica.
|
Notas: El nevus melanocítico intradérmico (Fig.12) corresponde a una lesión benigna muy frecuente, formada por melanocitos dentro de la dermis que forman una lesión solevantada en la piel. Melanoma (Fig.13): es un tumor maligno originado en las celúlas melanocíticas, con capacidad de dar metástasis a distancia. |
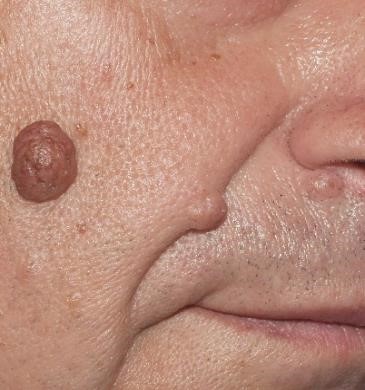
Figura 12: Nevos melanociticos intradérmicos faciales.
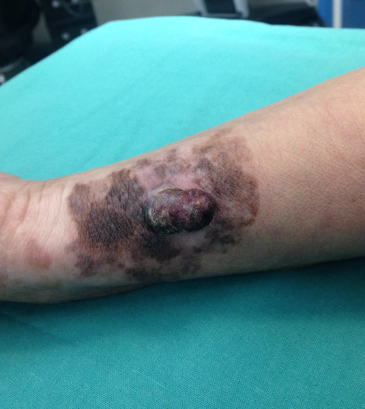
Figura 13: Melanoma antebrazo.
Célula de Langerhans
Células dendríticas mononucleares que actúan como presentadores de antígenos en la activación de linfocitos T, iniciando la respuesta inmune, que es uno de los principales mecanismos de daño en dermatología. Se encuentran dispersas entre los queratinocitos, principalmente en el estrato espinoso. En la microscopía tradicional no son visibles con técnicas convencionales, sólo con técnicas de inmunohistoquímica (Fig.14) pero con microscopía electrónica se observan los gránulos de Birbeck,característicos de estas células (Fig.15).
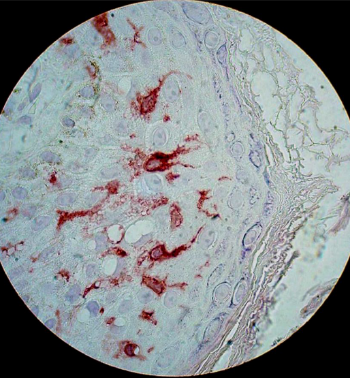
Figura 14: Células de Langerhans con tinción inmunohistoquímica.
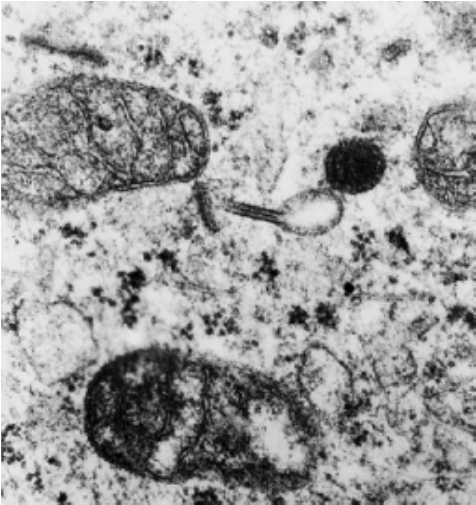
Figura 15: Microscopía electrónica: Gránulos de Birbeck
Células de Merkel
Células no dendríticas que se ubican en estrato basal de la epidermis. Son difíciles de identificar en microscopía convencional. Son mecanorreceptores ubicados en sitios de alta sensibilidad táctil como la cara y los dedos. Existen tumores asociados a estas células que son muy malignos y poco frecuentes (Fig.16).
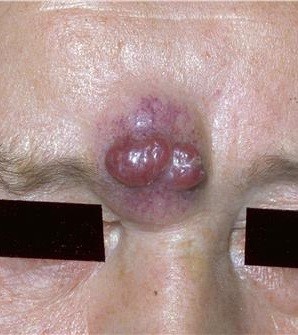
Figura 16: Tumor de células de Merckel.
Unión Dermoepidérmica (Fig.17):
Zona que une la epidermis (epitelio) con la dermis (tejido conectivo) y que cumple importantes roles para el funcionamiento de la piel:
- Cohesión entre epidermis y dermis
- Sostén para epidermis
- Determina polaridad del crecimiento
- Dirige organización del citoesqueleto de las células basales
- Proporciona señales para sucesos morfogénicos
- Barrera semipermeable
La unión dermoepidérmica se puede dividir en 4 porciones:
- Lámina basal del epitelio
- Lámina lúcida
- Lámina densa
- Lámina reticular
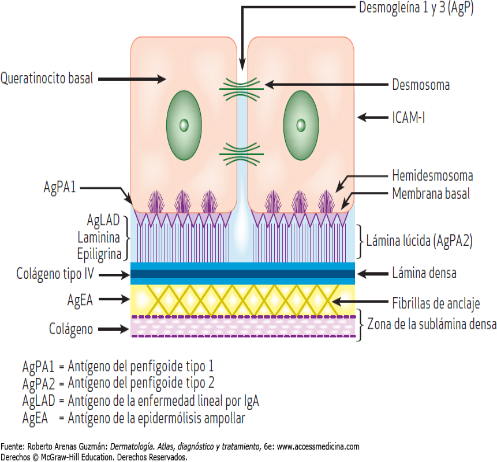
Figura 17: Esquema Unión dermoepidérmica.
Notas:
- En la unión dermoepidérmica se pueden producir enfermedades que al atacar este anclaje entre los dos tejidos lleva a la formación de ampollas. Las principales enfermedades que tienen la unión dermoepidérmica como blanco son:
- Penfigoide ampollar: alteración inmunológica mediada por anticuerpos dirigidos contra hemidesmosomas entre los queratinocitos y la membrana basal. Se caracteriza por grandes bulas de contenido seroso y serohemático, tensas y distribuidas por todo el cuerpo. Se presenta generalmente en personas mayores.
- Epidermolisis bulosa de la unión: patología en que se generan ampollas al mínimo roce. Es lo que popularmente se conoce como piel de cristal.
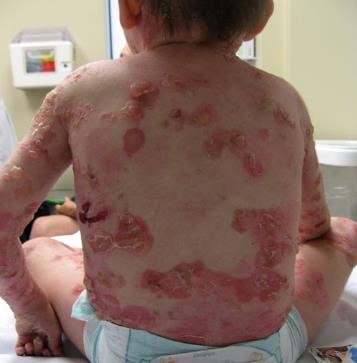
Figura 18: Paciente con penfigoide ampollar,zona muslos.
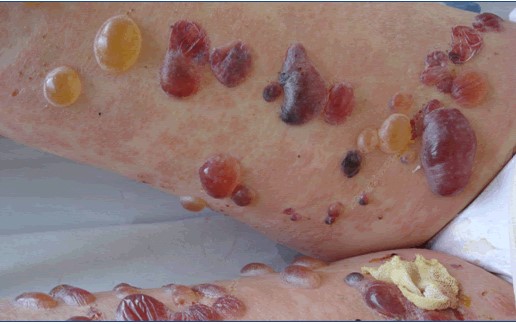
Figura 19: Epidermolisis bulosa.
B. Dermis (Fig.20)
Tejido conectivo constituido por células , una matriz extracelular (MEC) fibrilar y una MEC no fibrilar o amorfa; además de anexos cutáneos, terminaciones nerviosas y un componente vascular (recordar que epidermis es avascular y se nutre por difusión desde la dermis).
Sus principales funciones son:
- Constituye un soporte para la piel
- Proporciona flexibilidad y resistencia a la tensión
- Protege de lesiones mecánicas
- Fija agua
- Ayuda en la regulación térmica
- Incluye receptores a estímulos sensoriales
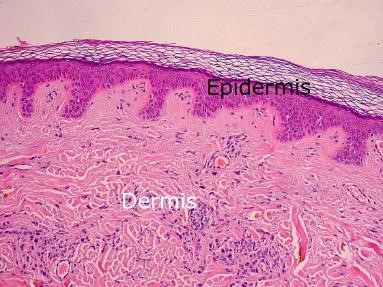
Figura 20: Epidermis y dermis.
Componentes de la dermis:
- MEC fibrilar: constituida por fibras colágenas, fibras reticulares y fibras elásticas.
- MEC no fibrilar o amorfa: conformada por glicosaminoglicanos (GAG) y Proteoglicanos, que permiten fijar agua en hasta 1.000 veces su volumen, proceso fundamental para mantener las características físico-químicas de la piel, y que clínicamente se manifiesta como el turgor y la elasticidad de esta.
-
<li><span style="text-decoration: underline;»>Componente celular:
- Células fijas o primarias: células propias de un tejido conectivo como la dermis. Incluye fibroblastos (principal célula del tejido), histiocitos o macrófagos fijos y células mesenquimáticas.
- Células agregadas o secundarias: células que se alojan en la dermis como sitio secundario (no el principal). Se incluye en este grupo mastocitos, plasmocitos y melanocitos.
- Células migradas: no son propias de la dermis pero pueden invadirla y habitualmente lo hacen. Incluyen: linfocitos, plasmocitos, polimorfonucleares neutrófilos y eosinófilos.
Regiones de la dermis
Podemos distinguir dos grandes regiones en la dermis según su ubicación, densidad celular, patrones de inervación, irrigación y organización del tejido conectivo:
- Dermis papilar: dermis superficial, correspondiente a las papilas, y se caracteriza por tener los vasos sanguíneos de los que saldrán nutrientes para difundir y llegar a la epidermis.
- Dermis reticular: dermis profunda, es menos celular que la papilar.
Vascularización
La dermis es un tejido bien irrigado, en el que encontramos distintos plexos y vasos:
- Arteriolas y vénulas principales
- Plexos vasculares: los más destacados son los plexos subpapilar, del folículo piloso y de las glándulas ecrinas.
- Glomus: estructura que hace de conexión entre las circulación arterial y venosa.
- Vasos linfáticos
Nota: El tumor glómico (Fig.21): es un tumor benigno,vascular,que se forma a partir del glomus. Su principal ubicación es subungueal, en los dedos de las manos, donde causa importante dolor y muchas veces alteraciones en la placa ungueal al comprimir la matriz ungueal.
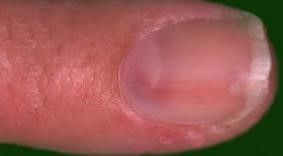
Figura 21: Tumor glómico.
Inervación
En la dermis encontramos importantes estructuras nerviosas encargadas de aspectos somatosensoriales (tacto, dolor, propiocepción, temperatura) y fibras eferentes autonómicas que regulan la vascularización, la piloerección y la secreción del sudor. Las estructuras más importantes son:
Figura 22: Hipodermis.
|
Funciones de la hipodermis |
|
Aisla el cuerpo Reservorio de calorías Barrera térmica Amortigua y protege a la piel Permite movilidad sobre estructuras adyacentes |
Notas:
- Ejemplo de patologías que se originan en la hipodermis son:
- Eritema nodoso (Fig.23): paniculitis septal, clínicamente son nódulos dolorosos principalmente en cara anterior de piernas.
- Celulitis (Fig. 24): infección del tejido celular subcutáneo.
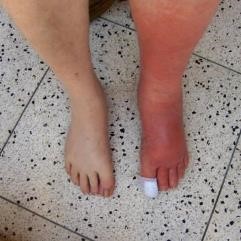
Figura 23: Celulitis pierna.
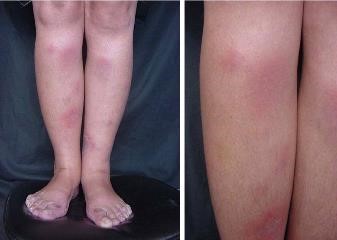
Figura 24: Eritema nodoso piernas.
