Generalidades
Las anemias que se asocian con eritrocitos normocíticos y normocrómicos, junto a una respuesta de reticulocitos muy baja (índice reticulocitario <2,0 a 2,5) son las anemias hipoproliferativas. Esta categoría comprende la ferropenia temprana (antes de que se produzcan eritrocitos hipocrómicos y microcíticos), la inflamación aguda y crónica, la insuficiencia renal, los estados hipometabólicos (desnutrición proteica y deficiencias endocrinas) y las anemias por lesión medular (aplásica [puede ser macrocítica], mieloptísica).
Las anemias hipoproliferativas son las anemias más comunes en clínica, estando en primer lugar la ferropénica seguida de la anemia por inflamación o enfermedades crónicas. Esta anemia, como la ferropénica, guarda relación en parte con un metabolismo anómalo del hierro. Las anemias que acompañan la insuficiencia renal, la inflamación, el cáncer y los estados hipometabólicos se caracterizan por una respuesta subóptima de la eritropoyetina a la anemia.
ANEMIA APLÁSICA
Definición y epidemiología
La anemia aplásica es una enfermedad de la médula ósea que se caracteriza por la presencia de una disminución del tejido hematopoyético, en ausencia de tumor, fibrosis u otros procesos como granulomas en la médula ósea, y que se acompaña de disminución de células sanguíneas en sangre periférica (una, dos o las tres series). En la práctica se debe sospechar anemia aplásica ante una pancitopenia y disminución de reticulocitos en sangre periférica. La médula ósea es hipocelular (menos de 25% de celularidad), lo que se confirma mediante biopsia de médula. Sin embargo, hay que tener presente que existen otras patologías que cursan con pancitopenia e hipocelularidad en la médula ósea (Ver Diagnóstico).
Es una enfermedad rara con una incidencia de 2 por millón de habitantes por año. Esta incidencia es 2 a 3 veces más alta en Asia del Este. En Chile se producen 2 a 4 casos nuevos al año. Tiene una distribución bifásica con un peak entre los 10-25 años y otro peak en los mayores de 60 años.
Etiología
Puede ser congénita o adquirida. La causa más frecuente de anemia aplásica es la idiopática.
a) Aplasias congénitas: las más raras.
- Anemia de Fanconi:
Enfermedad que suele manifestarse en la infancia entre los 5 y 10 años. Se caracteriza por la presencia de anomalías cromosómicas en los linfocitos de sangre periférica o las de la médula ósea. Se trata de un trastorno hereditario con carácter autosómico recesivo.
Manifestaciones: anemia, malformaciones fundamentalmente cutáneas (manchas café con leche) y óseas (hipoplasia del pulgar y malformación del radio). Malformaciones menos frecuentes: renales, oculares, microcefalia, sordera o retraso mental.
- Disqueratosis congénita:
Mucho más infrecuente que la anemia de Fanconi. Tiene una transmisión ligada al cromosoma X. También presenta alteraciones cutáneas asociadas. Se caracteriza por la tríada de leucoplasia en membranas mucosas, uñas distróficas e hiperpigmentación reticular.
- Aplasias selectivas congénitas:
Lesión de la médula ósea que afecta una sola serie hematopoyética.
- Síndrome de Diamond-Blackfan: aplasia pura de células rojas (eritroblastopenia). Se caracterizan por la casi ausencia de reticulocitos en la sangre periférica.
- Síndrome de Schwachman: aplasia pura de células blancas (agranulocitosis congénita). Se acompaña de insuficiencia exocrina del páncreas.
- Síndrome de Kostmann: aplasia pura de células blancas (agranulocitosis congénita).
- Trombocitopenia amegacariocítica: aplasia pura de megacariocitos.
b) Aplasias adquiridas: las más frecuentes.
Dentro de ellas se puede distinguir:
- Primarias. La mayoría (hasta el 50% de los casos) son de causa desconocida o idiopáticas.
- Secundarias. Ver Tabla 1.
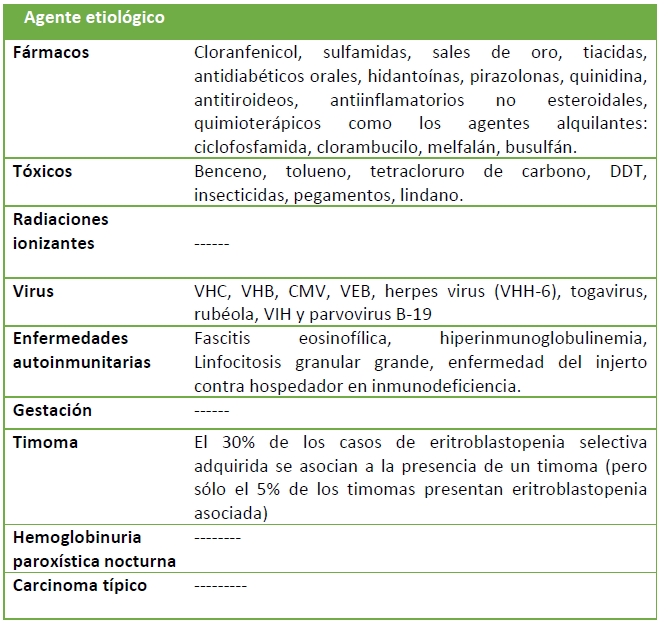
Tabla 1. Causas más frecuentes de aplasias adquiridas secundarias.
Fisiopatología
Existen tres hipótesis para explicar la lesión medular en la aplasia:
- Defecto intrínseco de las células germinales de la médula ósea.
- Defecto del denominado microambiente de la médula ósea (tejido vascular y conjuntivo de soporte).
- Anomalías en la regulación inmunológica (humoral y/o celular) de la hematopoyesis.
La mayoría de los casos adquiridos son secundarios a un proceso inmunitario por expansión oligoclonal de células T citotóxicas que secretan IF-γ y TNF-α causanando la muerte de células hematopoyéticas por apoptosis.
La falla de la médula ósea se debe al daño grave al comportamiento de las células hematopoyéticas. En la anemia aplásica resulta evidente el reemplazo de la médula ósea por grasa en la muestra de biopsia y en la resonancia magnética de la columna vertebral. Las células que portan el antígeno CD34, marcador de las células hematopoyéticas tempranas, están muy disminuidas y en los estudios funcionales se advierte la ausencia de células progenitoras comprometidas y primitivas. Las pruebas in vitro sugieren que la reserva de células madre se reduce a ≤1% de lo normal en la enfermedad grave al momento de la presentación.
Existe un defecto intrínseco en las células madre en las anemias aplásicas constitucionales: las células de los pacientes con anemia de Fanconi tienen daño cromosómico y sufren muerte con la exposición a ciertos agentes químicos. Los telómeros son cortos en algunos pacientes con anemia aplásica por mutaciones heterocigóticas en genes del complejo reparador del telómero. Los telómeros también pueden acortarse por mecanismos fisiológicos en la falla medular adquirida por las demandas de la replicación sobre una reserva limitada de células madre.
Diagnóstico
La clínica de la aplasia medular deriva de la disminución de las células sanguíneas de las tres series hematopoyéticas. De esta forma, se presenta con síndrome anémico, infecciones de repetición como consecuencia de la neutropenia y fenómenos hemorrágicos por la trombopenia. Si bien desde el punto de vista práctico conviene pensar en aplasia cuando un paciente presenta pancitopenia, esta puede ocurrir en otras enfermedades, como muestra la Tabla 2.
La anemia aplásica puede aparecer de manera súbita o insidiosa. La hemorragia es el síntoma temprano más frecuente; existen proclividad a la equimosis días o semanas antes, rezumamiento de las encías, epistaxis, sangrado menstrual abundante y en ocasiones petequias. En la trombocitopenia es inusual la hemorragia masiva, pero el sangrado leve en el sistema nervioso central pueden provocar hemorragia intracraneal o retiniana catastrófica. También son frecuentes los síntomas de anemia, entre ellos laxitud, debilidad, disnea y una sensación de golpeteo en los oídos. La infección es inusual como primera manifestación en la anemia aplásica (a diferencia de la agranulocitosis, en la que la faringitis, la infección anorrectal o la septicemia franca ocurren pronto). Una característica notoria de la anemia aplásica es la limitación de los síntomas al sistema hematológico; a menudo, los pacientes no sienten molestia alguna a pesar del descenso drástico de las cifras de biometría hemática. El malestar general y la pérdida ponderal deben señalar otras causas de pancitopenia. Con frecuencia se necesita la anamnesis repetida para identificar el uso previo de fármacos, exposición a químicos y enfermedades virales precedentes. Un antecedente familiar de enfermedades hematológicas o trastornos sanguíneos; fibrosis pulmonar o hepática, o encanecimiento temprano indica una telomeropatía.
En el frotis sanguíneo se observan eritrocitos grandes y escasez de plaquetas y granulocitos. A menudo aumenta el volumen corpuscular medio (VCM). Los reticulocitos están disminuidos o ausentes y la cifra de linfocitos puede ser normal o baja.
El diagnóstico de la anemia aplásica casi siempre es sencillo y se basa en la combinación de pancitopenia y médula ósea adiposa. Ver Figura 1.
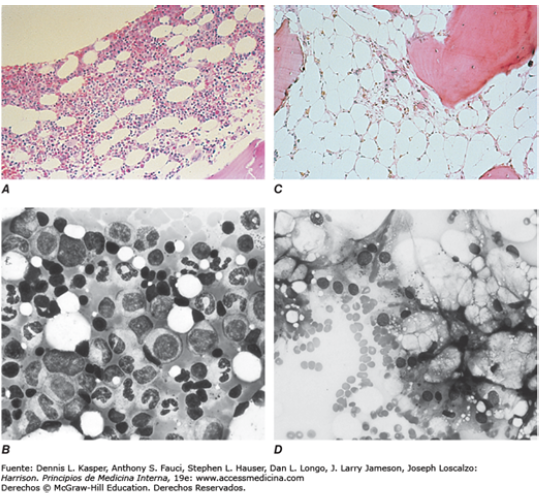
Figura 1. Médula ósea normal y aplásica. A: Biopsia de médula ósea normal. B: Frotis de aspirado medular normal. La médula ósea tiene 30 a 70% de células y muestra una mezcla heterogénea de células mieloides, eritroides y linfoides. C: Biopsia en anemia aplásica. D: Frotis medular en la anemia aplásica. La médula muestra reemplazo de tejido hematopoyético por grasa y sólo se observan células estromales y linfoideas residuales.
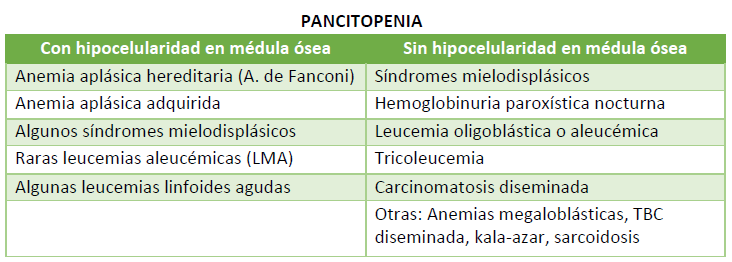
Tabla 2. Diagnósticos diferenciales de pancitopenia con o sin hipocelularidad en médula ósea.
Criterios diagnósticos para la anemia aplásica:
1. Disminución de celularidad en médula ósea menor al 25% de lo normal.
2. a) Ausencia de mielodisplasia (diagnóstico por morfología y estudio citogenético de la médula ósea).
b) Hemoglobinuria paroxística nocturna (estudio por citometría de flujo en sangre periférica).
c) Enfermedad de Fanconi (ausenta de fragilidad cromosómica en sangre periférica).
Criterios de gravedad de la anemia aplásica:
1. Moderada:
- Hipocelularidad de la médula ósea inferior al 30%
- Ausencia de pancitopenia grave
- Disminución de al menos dos de las tres series por debajo de lo normal
- RAN* > 500 /mm3
2. Grave:
- Hipocelularidad de la médula ósea inferior al 25%
Además de dos de los siguientes parámetros:
- RAN < 500/ mm3
- Trombopenia < 20.000/ mm3
- Reticulocitos < 1% (corregido por el hematocrito)
3. Muy grave:
Si se cumplen los criterios para anemia aplásica grave y:
- RAN < 200/ mm3
*RAN: Recuento absoluto de neutrófilos.
Diagnóstico diferencial
La presencia de esplenomegalia, casi siempre descarta el diagnóstico de aplasia, y debería orientar hacia otras patologías como la hepatopatía grave, tricoleucemia, mielofibrosis con metaplasia mieloide, policitemia vera en fase gastada, kala-azar, enfermedad de Gaucher, Síndrome de Banti.
Siempre tener presente las otras causas de falla medular como el reemplazo medular por células leucémicas, linfoma, cáncer y fibrosis. La hematopoyesis megaloblástica por déficit de vitamina B12 o ácido fólico, la hemoglobinuria paroxística nocturna, los Síndromes mielodisplásicos (SMD) y finalmente la infección por VIH y el Síndrome Hemofagocítico viral.
Tratamiento
Dependerá de la gravedad de la anemia aplásica, la disponibilidad de un donante familiar histocompatible y la edad del paciente.
Aplasia grave y muy grave:
- Tratamiento general: Todos los pacientes deben recibir tratamiento de soporte con transfusión de glóbulos rojos y/o plaquetas, además del manejo de la neutropenia febril, que muchas veces es la principal causa de muerte.
- Tratamiento específico:
- Menores de 40 años: Trasplante alogénico de progenitores hematopoyéticos, si tiene donante familiar idéntico. Las tasas de respuesta son cercanas al 80%.
- Mayores de 40 años y menores sin donantes: Tratamiento inmunosupresor con globulina antitimocito o antilinfocitaria, ciclosporina y prednisona. Las tasas de respuesta ascienden al 60-70%. La mejoría se observa después de tres meses de recibida la infusión de timoglobulina. La ciclosporina debe prolongarse mínimo un año.
*Otros tratamientos son:
- Esteroides en dosis altas en eritroblastopenia congénita.
- Citostáticos como la ciclofosfamida.
- Andrógenos en el caso de aplasias leves (como efectos secundarios: ictericia colestásica y hepatocarcinoma).
- El tratamiento del parvovirus B-19 suele ser gammaglobulina intravenosa.
- Factores de crecimiento hematopoyético.
Resumen: Aspectos claves
- Debe sospecharse anemia aplásica con una clínica derivada de la pancitopenia: síndrome anémico, infecciones y hemorragias.
- El diagnóstico se obtiene mediante el estudio de médula ósea, que será hipocelular (<25%). No hay esplenomegalia.
- La causa más frecuente de anemia aplásica es la idiopática.
- Su tratamiento será el transplante alogénico de progenitores hematopoyéticos de donante familiar. Sin embargo, en mayores de 40 años o en menores sin donantes compatibles se indicará tratamiento inmunosupresor con globulina antitimocito (sus efectos se aprecian luego de 3 meses), ciclosporina (por un año) y prednisona.
Caso clínico
ANAMNESIS PRÓXIMA:
Motivo de ingreso: Síndrome anémico
Paciente de sexo femenino de 18 años, consulta por cuadro clínico de 6-8 meses de evolución caracterizado por astenia progresiva, fatiga en relación a actividades habituales, palidez progresiva y disnea de moderados esfuerzos. Último mes con metrorragia persistente asociada a dolor tipo cólico en el hipogastrio. Ingresa a Ginecología, donde le realizan una Ecografía ginecológica, la que resulta normal. Su hemograma muestra pancitopenia. Se transfunden glóbulos rojos y se deriva a Hematología para estudio y tratamiento.


1. Bases de la Medicina Clínica. Unidad 15: Hematología. Tema 15.2: Anemia por enfermedad crónica.
2. Hematología. Manual CTO de Medicina y Cirugía. 1era edición. Chile.
3. Principios de Medicina Interna. Harrison. McGraw Hill. 19a edición. 2015.
