Discrasias de células plasmáticas
Generalidades
-
Definición: La discrasias de células plasmáticas se entienden como neoplasias monoclonales relacionadas entre sí debido a que proceden de progenitores comunes pertenecientes a la línea celular de los linfocitos B.
-
Cuadros clínicos que componen el síndrome o conjunto de patologías, idealmente ordenados de acuerdo a los criterios mas utilizados: Las discrasias de células plasmáticas son:
Mieloma Múltiple (MM), la macroglobulinemia de Waldenström, la amiloidosis primaria, y las enfermedades de las cadenas pesadas.
-
Epidemiología global del síndrome o conjunto de patologías:
La incidencia del mieloma alcanza su máximo en los estadounidenses de raza negra y habitantes de las islas del Pacífico; la cifra es intermedia en europeos y sujetos de raza blanca estadounidenses y mínima en países en desarrollo, incluidos los de Asia.
Mieloma Múltiple
-
Definición y epidemiología
El mieloma múltiple se define como una proliferación maligna monoclonal de células plasmáticas. El tumor, sus productos, y la respuesta del hospedero generan varios trastornos funcionales, orgánicos y síntomas. Por ejemplo: Dolor óseo, fracturas, lesiones óseas osteolíticas, insuficiencia renal, anemia, hipercalcemia, y predisposición a infecciones.
El MM representa el 1% entre el cáncer y el 10%-13% del cáncer hematológico. Edad de presentación promedio es de 70 años. La incidencia en Chile es de 2,9 casos/100.000 habitantes en hombres y de 2,5 en la mujer. La incidencia ha aumentado en el país y tanto esta como la prevalencia aumentan con la edad. El pronóstico de los pacientes es variable.
-
Etiología
Se desconoce la etiología del MM, aunque se ha observado con más frecuencia entre campesinos, carpinteros, curtidores de piel y los que tienen contacto con productos del petróleo. También en personas expuestas a radiación nuclear. En los individuos se han encontrado alteraciones cromosómicas con importancia pronóstica (deleciones de 13q14 y de 17p13) así como translocaciones en t(11;14) (q13;q32) y t(4;14)(p16;q32)
-
Fisiopatologia
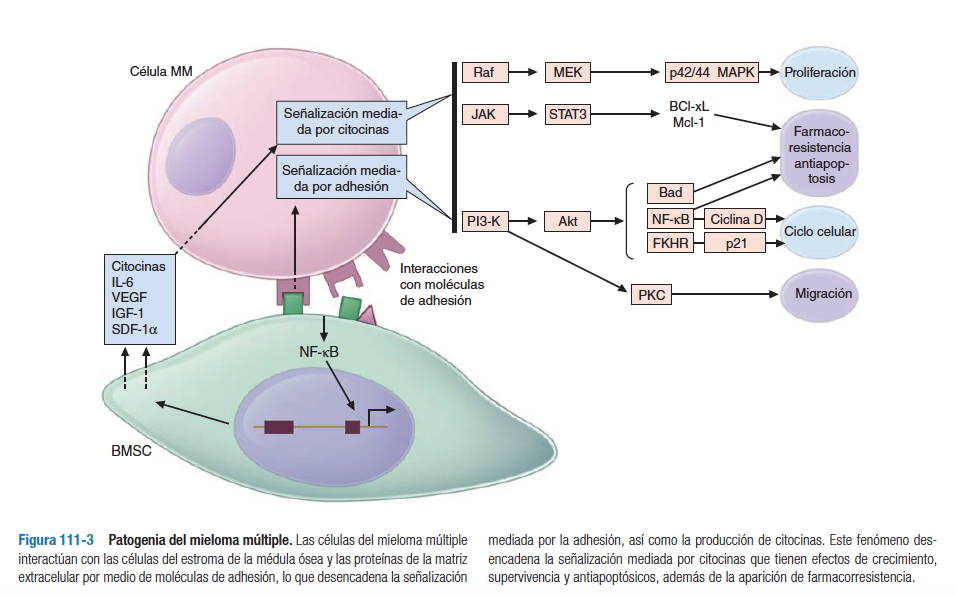
Mutaciones genéticas alteran la expresión de moléculas de adhesión en las células del MM, así como la respuesta a estímulos de crecimiento en el microambiente.
Interacciones entre las células del MM y de la MO o proteínas de la MEC están mediadas por: receptores de membrana (Integrinas, cadherinas, selectinas, y moléculas de adhesión) y tienen efectos en el crecimiento tumoral, supervivencia tumoral, migración y resistencia a drogas.
La adhesión de las cél del MM a las células estromales y hematopoyéticas induce la secreción de citoquinas y factores de crecimiento. Incluyendo IL-6, VEGF, IGF-1, miembros de la familia TNF, factor de crecimiento transformados β1, y IL-10. Estas citoquinas y factores de crecimiento son producidos y secretados por células en la MO, incluyendo aquellas del MM. Y son reguladas de manera autocrina y paracrina.
La adhesión de las cél del MM a células de la MEC ( colágeno, fibronectina, laminina y vitronectina) desencadena el “up-regulation” de proteínas reguladoras del ciclo celular y de proteínas antiapoptóticas. Las lesiones óseas son causadas por un desbalance entre los osteoblastos y osteoclastos.
La inhibición de la vía de Wnt suprime los osteoblastos, mientras que la amplificación de la vía de RANK y la acción de MIP1α (macrophage inflammatory protein) activa los osteoclastos.
La inducción de moléculas proangiogénicas (VEGF), aumenta la densidad microvascular de la médula ósea y da cuenta de la estructuna anormal de los vasos sanguíneos en el tumor de MM.
-
Clasificación


-
Clínica
Del 20 a 30% de los pacientes son asintomáticos y su diagnóstico es casual (al encontrar velocidad de sedimentación globular elevada, anemia leve o presencia de paraproteína en la sangre)
-
Enfermedad ósea: aparición de lesiones osteolíticas (por la proliferación de células tumorales y destrucción del hueso por la actividad aumentada de osteoclastos), principalmente en huesos hematopoyéticos (cráneo, costillas, vértebras,pelvis y epífisis de huesos largos). El dolor óseo es el síntoma más frecuente (70% de los pacientes), se manifiesta como dolor de espalda o costillas que aumenta al movimiento. Si el dolor es persistente y localizado, hay que sospechar fractura patológica. Pueden no haber lesiones osteolíticas y encontrarse una osteoporosis difusa (Dg. Dif; osteoporosis de origen desconocido).
Como consecuencia de las lesiones óseas puede haber compresión radicular o medular por aplastamiento vertebral.
-
Infecciones: Consecuencia de la alteración de la inmunidad humoral, disminución de la concentración de Ig’s normales, y del tratamiento con corticoides y quimioterápicos, aumenta el riesgo de infección (mayoritariamente de gérmenes encapsulados, neumonía y pielonefritis, por Streptococcus pneumoniae, Staphilococcus aureus, Klebsiella pneumoniae, Escherichia coli y gramnegativos)
-
Afectación renal: Insuficiencia renal posible por:
-
Hipercalcemia: produce hipercalciuria y diuresis osmótica, que genera depleción de volumen y falla renal. También pueden haber depósitos de calcio que generen nefritis intersticial
-
Excreción de cadenas ligeras: Es la causa más frecuente de IR en MM. Hay proteinuria de Bence-Jones, cuyo mecanismo de nefrotoxicidad es desconocido.
-
Otras: hiperuricemia, amiloidosis, pielonefritis a repetición, síndrome de hiperviscosidad, consumo de AINE e infiltración al riñón por cél. Plasmáticas.
-
Insuficiencia de médula ósea: Se produce anemia por la ocupación de la MO por las cél plasmáticas
-
Hipercalcemia: hasta en el 30%. Que produce síntomas como anorexia, nauseas, vómitos, poliuria, polidipsia, estreñimiento y confusión.
-
Hiperviscosidad: muy poco frecuente (en MM igM e IgG3), se presenta con alteraciones neurológicas, visuales (fondo de ojo con venas tortuosas y dilatadas), alteraciones hemorrágicas, insuficiencia cardíaca y circulatoria.
La valoración clínica de los pacientes con mieloma incluye una exploración física cuidadosa en busca de dolor óseo a la palpación y tumoraciones.
-
Diagnóstico
La tríada clásica del mieloma incluye plasmocitosis medular (>10%), lesiones osteolíticas y un componente M en el suero, la orina, o ambos.
-
Criterios de Swog:
C. Mayores:
a) Plasmocitoma en biopsia tisular
b) Células plasmáticas en MO superiores al 30%
c) Pico monoclonal sérico >3,5 g/dl si es IgG
Pico monoclonal sérico > 2 g/dl si es IgA
Proteinuria de cadenas ligeras > 1g/día
C. Menores:
1) Celularidad plasmática en MO entre 10 y 30%
2) Pico monoclonal inferior al criterio mayor
3) Lesiones osteolíticas radiológicas
4) Disminución de Ig normales
Se considera diagnóstico de mieloma cuando se reúne el criterio A o B con criterios menores, o cuando existe aisladamente el criterio C, o cuando se reúnen criterios menores entre sí.
-
Criterios de Kyle:
-
Más del 10% de células plasmáticas en MO
-
Demostración de plasmocitoma
-
Componende M en suero IgG >3 g/dl, IgA>2 g/dl, cadenas ligeras en orina >1g en 24 hrs, lesiones osteolíticas.
Se considera diagnóstico de mieloma: se exige presencia de más del 10% de cél plasmáticas en MO, o demostración de plasmocitoma más uno de los siguientes:
-
Componente en suero IgG >3g/dl, IgA >2g/dl
-
Cadenas ligeras en orina >g/ 24hrs
-
Lesiones osteolíticas
Estadificación del MM (Se ha reemplazado por el ISS)

** Extra: según Harrison:
Las radiografías de tórax y huesos pueden indicar la presencia de lesiones líticas u osteopenia difusa. Las imágenes por resonancia magnética (MRI, magnetic resonance imaging) son un método sensible para corroborar la compresión de médula o raíces nerviosas en individuos con síndromes dolorosos. En las biometrías hemáticas completas puede encontrarse anemia, y la velocidad de eritrosedimentación está elevada.
Un número muy escaso (cerca del 2%) de pacientes tiene leucemia de células plasmáticas, con >2 000 células/μl. Esto ocurre con una frecuencia desproporcionadamente alta en los mielomas IgD (12%) e IgE (25%).
Puede haber concentraciones séricas altas de calcio, nitrógeno ureico, creatinina y ácido úrico. La electroforesis de las proteínas y la medición de las inmunoglobulinas séricas sirven para detectar y caracterizar los picos M, junto con la inmunoelectroforesis, que es muy sensible para identificar las concentraciones bajas de los componentes M que no se descubren con la electroforesis de las proteínas. Es necesario analizar una muestra de orina de 24 h para medir la excreción de proteínas Bence Jones. La fosfatasa alcalina sérica suele ser normal a pesar de la extensa afectación ósea, debido a la falta de actividad osteoblástica. También es importante cuantificar la microglobulina β2 sérica.
-
Tratamiento:
-
En etapa inicial, asintomática, puede no tratarse ya que no prolonga la supervivencia. El mieloma latente tampoco requiere tratamiento.
-
Cuando es sintomático hay que comenzar tratamiento quimioterápico.
-
Pacientes mayores a 70 años que no van a recibir autotransplante , melfalán o ciclofosfamida con prednisona. Se debe mantener al menos un año en pacientes que hayan conseguido el plateau, el tratamiento de mantención es talidomisa o esteroides.
-
En pacientes menores a 70 años: poliquimioterapia como bortezomib (inhibidor proteasoma) más dexametasona en dosis altas para pasar a la fase de consolidación con autotransplante de progenitores hematopoyéticos.
RAM: La talidomida trombosis y neuropatía periférica.
Lenalidomida: trombosis y citopenias
Bortezomib: neuropatía periférica y diarrea
En la talidomida y lenalidomida se debe iniciar el tratamiento profiláctico con AAS o HBPM por el alto riesgo trombótico.
Se considera remisión completa la ausencia de paraproteína en suero y orina, determinada con inmunofijación y mantenida por 6 semanas, desaparición plasmocitoma, existencia de menos de un 5% de cél plasmáticas en la MO y la estabilización del número y tamaño de lesiones osteolíticas.
-
Pronóstico y Seguimiento:

