Nivel de manejo del médico general: Diagnóstico Sospecha Tratamiento Inicial Seguimiento Derivar
Aspectos esenciales
- Se habla de grave cuando hay un recuento bajo 20.000 plaquetas /mm3, pues hay sangrado espontáneo
- Se presenta con equimosis, petequias y hemorragias.
- Los mecanismos son por déficit de producción o aumento de la destrucción.
- El tratamiento dependerá de la alteración basal.
Caso clínico tipo
Paciente que consulta por hematuria y hematomas en su cuerpo sin relación con traumatismos. El recuento de plaquetas es de 30.000/mm3.
Definición
La trombocitopenia se define como un recuento de plaquetas por debajo del límite inferior de la normalidad (es decir, <150.000/microL para los adultos). Los grados de la trombocitopenia pueden subdividirse en leve (recuento de plaquetas de 100.000 a 150.000/microlitro), moderada (50.000 a 99.000/microlitro) y severa ( <50.000/microlitro).
La trombocitopenia grave confiere un mayor riesgo de sangrado, pero la correlación entre el recuento de plaquetas y el riesgo de sangrado varía de acuerdo a la condición subyacente y puede ser impredecible.
Etiología-epidemiología-fisiopatología
Las causas de trombopenia se pueden dividir en:
- Disminución de la producción de plaquetas, dentro de las cuales están: anemia aplástica, leucemia, etc.
- Destrucción o consumo de plaquetas: púrpura trombocitopénica idiopática (PTI) (causa auto-inmune y afectación de producción), VIH, coagulación intravascular diseminada (CID), púrpura trombocitopénica trombótica (PTT) y síndrome hemolítico urémico (SHU).
Diagnóstico
La historia debe centrarse en los recuentos plaquetarios previos, antecedentes familiares, sangrado, medicamentos, el exceso de medicamentos de venta libre, exposiciones infecciosas, las prácticas dietéticas, y otras condiciones médicas (por ejemplo, trastornos hematológicos, condiciones reumatológicas, cirugía, transfusión).
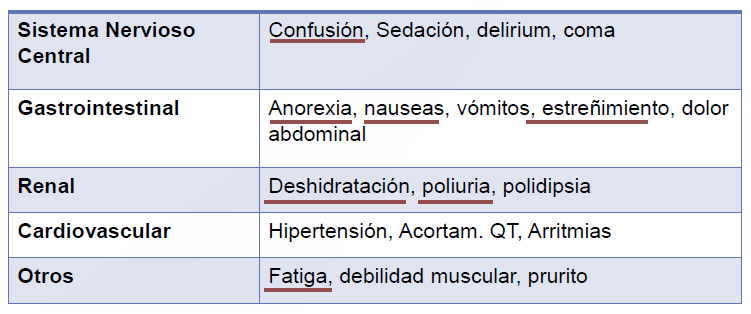
El examen físico debe evaluar el sangrado, ya que se presenta con sangrado cutáneo en forma de equimosis y petequias espontáneas, hemorragias nasal, gingival, menorragia e inclusive sangrado urinario o gastrointestinal. También debe buscarse linfadenopatía, hepatoesplenomegalia y trombosis.
PTI se presenta con síndromes purpúricos o una trombocitopenia aislada en los exámenes de sangre.
El SHU y la PTT presentan: anemia hemolítica microangiopática, trombocitopenia, fiebre, compromiso renal y compromiso neurológico.
Se debe buscar infecciones por virus o evaluar el consumo de heparina. Si hay sepsis asociado, pensar en CID.
No hay pruebas de laboratorio adicionales además del recuento plaquetario y frotis de sangre periférica que sean absolutamente necesaria en un paciente con trombocitopenia aislada. Los adultos con nueva trombocitopenia deben tener pruebas de VIH y VHC. Las pruebas adicionales de laboratorio puede estar justificada en pacientes con otros hallazgos.
La urgencia depende del grado de trombocitopenia y otras anomalías, y la estabilidad de los resultados.
La evaluación de la médula ósea no es necesaria en todos los pacientes con trombocitopenia. Sin embargo puede ser útil en algunos paciente si se sospecha de una enfermedad hematológica primaria.
Tratamiento
El manejo de pacientes con trombocitopenia depende del diagnóstico subyacente. Principios generales que se aplican a todos los pacientes incluyen evitar los medicamentos que interfieren con la función plaquetaria, coordinación con los anestesiólogos y cirujanos antes de procedimientos invasivos, y corrección de alteraciones de la coagulación. A menudo no se necesitan restricciones de actividades.
La terapia del PTI se inicia con prednisona 1mg/Kg o metilprednisolona según gravedad de Trombopenia. Si fracasa se puede utilizar Ig-ev o esplenectomía.
El SHU requiere plasmaféresis, infusión PFC y soporte renal.
El tratamiento del PTT es plasmaféresis, PFC y uso de esteroides. En caso de trombopenia causado por heparina, la suspensión de esta es crítica.
En los casos de CID, el tratamiento inicial es tratar la patología de base.
Transfusión de Plaquetas: Indicada en forma
– Terapéutica: hemorragia activa atribuible a trombocitopenia.
– Profiláctica (sin hemorragia activa): en patología médica con recuento <10.000/mm3; y en pacientes quirúrgicos y obstétricos con recuento <50.000/mm3.
– Contraindicado en PTI (a menos que tenga riesgo vital), PTT, PT Postransfusional
Seguimiento
Derivar a especialista para estudio etiológico.
Referencias
1) https://www.uptodate.com/contents/approach-to-the-adult-with-unexplained-thrombocytopenia?source=search_result&search=trombocitopenia%20severa&selectedTitle=1~150