GENERALIDADES
Los linfomas son neoplasias que se originan en las células del sistema inmune, estos derivan de células que normalmente se desarrollan en linfocitos T o linfocitos B. Afectan a células en distintas etapas de diferenciación, por lo que presentan una diversidad de datos morfológicos e inmunitarios, además de variadas manifestaciones clínicas.
Históricamente, las neoplasias de tipo linfoide que se presentan como complicaciones en la médula y la sangre (leucemias) han sido separadas de las que se presentan como una masa (linfomas). Sin embargo, ahora se precisará que cualquier «linfoma» puede presentarse con o evolucionar a un cuadro leucémico, y cualquier «leucemia» en ocasiones se puede presentar con una lesión de masa.
En la clasificación de la OMS, el diagnóstico de las diversas neoplasias linfoides no depende de la localización anatómica de las células tumorales, sino más bien en las características de la célula de origen del tumor, como se juzga por la morfología, inmunofenotipo, y los hallazgos genéticos. Dado a las diferencias que se pueden encontrar, los clasificaremos, a grandes rasgos, en Linfoma de Hodgkin (LH) (detallado en otro apunte) y en Linfoma no Hodgkin (LNH).
Hasta mediados del siglo XX eran enfermedades fatales, actualmente es posible curar aproximadamente el 80% de los linfomas de Hodgkin y el 50-60% de los linfomas no Hodgkin.
LINFOMA NO HODGKIN
1. DEFINICIÓN Y EPIDEMIOLOGÍA
Los linfomas no Hodgkin son neoplasias de origen linfoide tipo B, T y excepcionalmente NK. Cerca del 90% de los casos derivan de una alteración linfocitaria B, mientras que una pequeña porción proviene de linfocitos T, a diferencia de la infancia, donde esto se invierte predominando los de origen linfoide tipo T.
Como se menciona previamente, son proliferaciones malignas del tejido linfático que se diferencian del Linfoma de Hodgkin por una variedad de características clínicas e histológicas (a detallar más adelante).
La mayoría se presenta con adenopatías de crecimiento progresivo e indoloras, donde predominan las cervicales, axilares o inguinales. Sin embargo, cerca de un 30% puede presentarse en sitios extra-ganglionares, cuya ubicación más frecuente es digestiva.
Esta enfermedad es más frecuente en el sexo masculino (relación hombre/mujer de 1,5:1) y en edades avanzadas, presentando su mayor ocurrencia entre los 45 a 70 años, con una media de 56 años. Existe una mayor incidencia en personas con inmunosupresión congénita o adquirida, enfermedades autoinmunes (Sjogren, tiroiditis, enfermedad celíaca), asociada a infecciones virales (Epstein Barr, HTVL-1) o Helicobacter pylori.
El linfoma no Hodgkin tiene una incidencia mayor que el linfoma de Hodgkin, siendo casi 5 veces más frecuente, ya sea a nivel mundial o de la población nacional:
- Tasa Internacional de Linfoma no Hodgkin: 6,9 por cien mil hab.
- Tasa Internacional de Linfoma de Hodgkin: 1,2 por cien mil hab.
- Tasa Chilena estimada de Linfoma no Hodgkin: 5,6 por cien mil hab.
- Tasa Chilena estimada de Linfoma de Hodgkin: 1,5 por cien mil hab.
2. ETIOPATOGENIA
La etiología de la mayoría de los casos de LNH es desconocida. Sin embargo, hay evidencia que la estimulación antigénica prolongada por infecciones virales o bacterianas aumenta la probabilidad de desarrollo de linfoma.
Dentro de las asociaciones más frecuentes se encuentran:
- Pacientes con infección por virus de inmunodeficiencia humana (VIH) tienen un mayor riesgo de desarrollar linfomas.
- Pacientes con inmunodeficiencia adquirida por uso prolongado de drogas inmunosupresoras, en receptores de trasplante de órganos sólidos, tienen un riesgo aumentado para desarrollar linfomas.
- Niños con linfoma de Burkitt en África, tienen integrado el virus de Epstein-Barr (VEB) en las células malignas en el 90% de los casos.
- La leucemia/linfoma T del adulto, está asociada al retrovirus humano HTLV-1 en el 100% de los casos.
- Linfoma gástrico asociado a mucosas (MALT), está asociado a la infección por la bacteria Helicobacter pylori.
- Linfoma primario de derrames, está asociado al virus herpes HHV8
Además, se han identificado alteraciones citogenéticas recurrentes en algunos linfomas, que comprometen genes específicos:
- Linfoma folicular: t(14;18), gen de Ig y Bcl2.
- Linfoma del manto: t(11;14), gen Bcl1 y ciclina D1.
- Linfoma de Burkitt: t(8;14), gen c-Myc y gen de Ig.
- Linfoma anaplástico células grandes: t(2;5), gen Alk.
3. CLASIFICACIÓN
Algunos ensayos clínicos han separado subtipos histológicos de acuerdo al comportamiento clínico habitual de cada una de las neoplasias linfoides, que pueden ser más o menos separados en tres grupos, de la siguiente manera:
· Indolente – La supervivencia de los no tratados generalmente se mide en años. Los linfomas indolentes representan del 35% al 40% de los LNH. Los subtipos histológicos más comunes son el linfoma folicular, leucemia linfocítica crónica / linfoma linfocítico pequeño, algunos casos de linfoma de células del manto, linfoma de la zona marginal, linfoma linfoplasmocítico, micosis fungoideas y el linfoma esplénico de la zona marginal.
· Agresivo – La supervivencia de los no tratados generalmente se mide en meses. Cabe destacar que aproximadamente la mitad de los LNH son agresivos. Los subtipos más comunes incluyen el linfoma difuso de células B grandes, linfoma de células T periféricas, y linfoma de células grandes anaplásico.
· Altamente agresivo– La supervivencia de los no tratados se mide en semanas. Los linfomas altamente agresivos, como grupo, representan aproximadamente el 5%. Estas enfermedades son poco comunes, y pueden surgir a partir de células B o células T.
Clasificación de la OMS de los linfomas no Hodgkin (de acuerdo a su agresividad clínica)
| LINFOMAS INDOLENTES |
| Neoplasias de células B |
| Linfoma linfocitico pequeño /Leucemia linfocítica crónica de células B |
| Linfoma linfoplasmocítico |
| Mieloma de células plasmáticas |
| Leucemia de células vellosas |
| Linfoma folicular (Grado I y II) |
| Linfoma de la zona marginal |
| Linfoma de las células del manto (algunos casos) |
| Neoplasias de células T |
| Leucemia linfocítica granular de células T grandes |
| Micosis fungoideas |
| Leucemia prolinfocítica de células T |
| Neoplasias de células Natural Killer |
| Leucemia linfocitica granular de células NK grandes |
| LINFOMAS AGRESIVOS |
| Neoplasias de células B |
| Linfoma folicular (grado III) |
| Linfoma difuso de células B grandes |
| Linfoma de las células del manto (algunos casos) |
| Neoplasias de células T |
| Linfoma de células T periféricas |
| Linfoma de células grandes anaplásico. |
| LINFOMAS ALTAMENTE AGRESIVOS |
| Neoplasias de células B |
| Linfoma de Burkitt |
| Leucemia/Linfoma linfoblástico de precursores B |
| Neoplasias de células T |
| Leucemia de células T del adulto |
| Leucemia/Linfoma linfoblástico de precursores T |
4. CUADRO CLÍNICO
Como se menciona previamente, la presentación clínica del LNH varía enormemente dependiendo del tipo de linfoma y de las áreas de participación.
En los casos típicos, el LNH se caracteriza por la aparición de adenopatías indoloras, de crecimiento progresivo (especialmente cervicales, supraclaviculares o axilares) que no regresan con antiinflamatorios. A pesar de que es menos frecuente que en el LH, cerca del 40% de los pacientes pueden presentar síntomas generales o “B” (fiebre, sudoración o baja peso (>10% peso corporal)).
Generalmente, al examen físico, los ganglios son mayores a 2 cm, tienen más de 1 mes de evolución, son gomosos, móviles e indoloros y no se encuentran adheridos a la piel ni a planos profundos. Además, se puede consignar hepatoesplenomegalia, compromiso amigdalino, tumor mediastínico o abdominal.
Aproximadamente el 50% de los pacientes desarrollan la enfermedad extra nodal (de forma secundaria) durante el curso de su enfermedad, mientras que entre 10% y 35% de los pacientes tienen linfoma extra nodal primaria al diagnóstico.
De forma un poco más específica encontramos que:
· Los linfomas indolentes tienen un comienzo insidioso (debido a su lenta reduplicación), siendo asintomáticos o presentándose únicamente como adenopatías de crecimiento lento, hepatomegalia, esplenomegalia o citopenias. Debido a la menor cantidad de mitosis, se le confiere un pronóstico de vida media prolongada, sin embargo, es menos sensible a la quimioterapia.
· Los linfomas agresivos se presentan comúnmente de forma aguda o subaguda (debido a su rápida proliferación), con masas de rápido crecimiento, síntomas B (fiebre, sudoración nocturna y pérdida de peso), y / o niveles elevados de lactato deshidrogenasa en suero y ácido úrico. En estos casos es frecuente la diseminación extra linfática.
La historia natural de estos tumores muestra una variabilidad significativa de paciente a paciente. Presentaciones menos comunes incluyen erupciones en la piel, prurito, hipersensibilidad a picaduras de insectos o mordeduras, fatiga generalizada, malestar general, fiebre de origen desconocido, ascitis y derrames.
5.- MANEJO CLÍNICO:
Existe una guía AUGE al respecto (Guía clínica AUGE de Linfomas en personas de 15 años y más, 2013).
6.- DIAGNÓSTICO
El diagnóstico del Linfoma no Hodgkin se realiza por biopsia excisional de un ganglio o tejido comprometido. La punción aspirativa o biopsia con aguja (trucut) no son recomendables por dar material insuficiente para una clasificación adecuada.
La decisión de biopsia de un ganglio linfático depende de la situación clínica, características del paciente (edad, sexo), así como de la ubicación del nodo (s) involucrado. Independientemente si la adenopatía se encuentra localizada, regional, de forma generalizada (si otras pruebas de diagnóstico no son compatibles con otro diagnóstico) un ganglio linfático se debe considerar para biopsia si una o más de las siguientes características de ganglios linfáticos está presente:
● Tamaño significativo (generalmente >2 cm)
● La persistencia durante más de cuatro a seis semanas
● Aumento progresivo de tamaño
Los estudios de imagen son un componente clave de la evaluación para la clasificación de los pacientes con LNH y pueden ayudar en la selección de un sitio de la biopsia para el diagnóstico. La modalidad de imagen preferida para la estadificación de un paciente con LNH depende de la avidez por fluorodeoxiglucosa (FDG) del subtipo histológico. La tomografía por emisión de positrones integrado con la tomografía computarizada (PET/TC) se prefiere para la estadificación de los linfomas nodales FDG-ávidos, mientras que la TC solo es el preferido para histologías FDG-no ávidos.
El estudio de inmunohistoquímica adicional es indispensable para un diagnóstico preciso, ya que el tratamiento y pronóstico dependen de él. El inmunofenotipo de las células del linfoma se puede determinar por citometría de flujo realizada en suspensiones de células individuales no fijadas frescas o por inmunohistoquímica en secciones de tejidos congelados o frescos fijos.
La clasificación histológica aceptada mundialmente se basa en aquella descrita por la OMS e integra características clínicas, inmunológicas y moleculares. Existen diferencias en los subtipos histológicos en diferentes partes del mundo. Así, en el mundo occidental, Europa y USA predominan los linfomas de células B (80-90%), en Asia y países de menor desarrollo socioeconómico, aumentan los linfomas T (15-30%).
Con respecto a los estudios genéticos, las muestras frescas y en algunos casos los tejidos fijos pueden ser estudiados por anomalías citogenéticas, translocaciones cromosómicas, inmunoglobulina o reordenamientos del gen receptor de células T (TCR), análisis de la expresión génica, y secuenciación de ADN. Estos y otros estudios pueden ser pruebas de complemento importante cuando las muestras se obtienen por FNA, donde el material de diagnóstico limitada está disponible para la evaluación histológica convencional.
La estadificación clínica, basada en la clasificación de Ann Arbor por etapas, se usa con más frecuencia para describir la extensión del linfoma no Hodgkin en adultos. Los estadios por lo general se clasifican con números romanos del I al IV (1-4). A los linfomas que afectan a un órgano que está fuera del sistema linfático (un órgano «extranodal») se les añade la letra «E» (por ejemplo, etapa IIE), mientras que a los que afectan el bazo se les añade una S.
ESTADIFICACION DE ANN-ARBOR
| ETAPA I |
| I.- El linfoma se encuentra solamente en un área ganglionar o un órgano linfático. |
| IE.- Se encuentra solamente en un área de un órgano que está fuera del sistema linfático (extralinfático). |
| ETAPA II |
| II.- El linfoma está en dos o más grupos de ganglios linfáticos en el mismo lado (superior “1” o inferior “2”) del diafragma (músculo que separa el tórax del abdomen). |
| IIE.- Un órgano o localización extralinfática más una o más áreas ganglionares al mismo lado del diafragma. |
| ETAPA III |
| III.- Áreas ganglionares a ambos lados del diafragma. |
| III1.- Limitado a abdomen superior (ganglios portales, celíacos, esplénicos y bazo). |
| III2.- Ganglios linfáticos inferiores (paraaórticos, ilíacos, inguinales o mesentéricos) con o sin compromiso de ganglios superiores. |
| IIIE.- Órgano o localización extralinfática mas compromiso ganglionar a ambos lados del diafragma. |
| IIIS.- Compromiso esplénico con áreas ganglionares a ambos lados del diafragma. |
| ETAPA IV |
| IV.- Compromiso difuso o diseminado de uno o más órganos extra linfáticos, con o sin afectación ganglionar (p ej. Hígado, médula ósea u otros sitios extranodales no contiguos a los ganglios. |
Además del estadio se utilizan los siguientes términos:
- Enfermedad voluminosa o Bulky: hace referencia a los tumores en el tórax que son al menos del ancho de 1/3 del tórax o los tumores en otras áreas que son al menos 10 centímetros (alrededor de cuatro pulgadas) de ancho. Por lo general, se indica que el linfoma es voluminoso añadiendo la letra X a la etapa. La enfermedad voluminosa podría requerir tratamiento más intensivo.
- A vs. B: A cada etapa también se le podría asignar una A o B. La letra B se añade si una persona tiene cualquiera de los siguientes síntomas B: Pérdida de peso de más del 10% en los 6 meses previos al diagnóstico (sin hacer dieta), fiebre y sudoración profusa durante la noche.
Los exámenes necesarios para la etapificación clínica, son los mismos que para linfoma de Hodgkin:
Exámenes para la etapificacion clínica de linfomas Hodking y no Hodgkin
| Biopsia ganglionar | Inmunohistoquímica y marcadores moleculares |
| Laboratorio | Hemograma y VHS, pruebas hepáticas, cretinina, deshidrogenasa láctica, beta-2 microglobulina, calcemia, uricemia, pruebas de coagulación, proteinemia total y albúmina, electroforesis. |
| Serología | VIH, HTLV-1, VEB, VHB y VHC. |
| Citometría de flujo | Estudio inmunofenotipo de líquidos, sangre o médula ósea. |
| Imágenes | Radiografía de tórax, Escáneres de tórax, abdomen y pelvis (ganglios patológicos >2cm). |
| Biopsia medula ósea | Cresta ilíaca postero superior (2cm largo). |
| Evaluación cardiaca | Electrocardiograma, Ecocardiograma (>50 años). |
| PET/CT | Para evaluación de masas residuales después de quimioterapia. |
| Opcionales (según clínica) | Endoscopía digestiva alta y detección de Helicobacter pylorii, resonancia magnética, punción lumbar. |
7.- TRATAMIENTO
El tratamiento de los linfomas no Hodgkin se basa en la quimioterapia, radioterapia y en los agentes biológicos como los anticuerpos monoclonales. La quimioterapia puede utilizarse como monodroga (1 sola droga) o más frecuentemente una combinación de drogas o poliquimioterapia.
Los linfomas son muy sensibles a la radioterapia, sin embargo, los linfomas no Hodgkin se consideran enfermedades diseminadas, por lo tanto su valor es limitado.
Los agentes biológicos o inmunoterapia como los anticuerpos monoclonales han demostrado aumentar el efecto de la quimioterapia, al usarlos asociados a la quimioterapia. El mejor ejemplo es el rituximab o anti CD20, que destruye linfocitos CD20 positivos, presente en los linfomas de estirpe B.
TRATAMIENTO DE LINFOMAS INDOLENTES:
A.- Etapas localizadas I – II (sobrevida global 80%)
– Radioterapia.
– Quimioterapia COP o R-COP.
B.- Etapas avanzadas III – IV (sobrevida global 60%)
– Asintomáticos: solo observación.
– Sintomáticos: Clorambulico > 70 años o R-COP < 70 años.
TRATAMIENTO DE LINFOMAS AGRESIVOS DE CÉLULAS B:
A.- Etapas localizadas I – II (sobrevida global 80%)
– Quimioterapia de 4 ciclos R-CHOP + RT localizada.
B.- Etapas avanzadas III – IV (sobrevida global 50%)
– Quimioterapia de 6-8 ciclos R-CHOP.
TRATAMIENTO DE LINFOMAS AGRESIVOS DE CÉLULAS T:
-Quimioterapia de 6-8 ciclos CHOP.
8.- PRONÓSTICO Y SEGUIMIENTO
Múltiples estudios han demostrado que el pronóstico del LNH es mucho más dependiente de la histopatología, siendo sólo secundariamente influenciada por parámetros clínicos como la edad, presencia de enfermedad extranodal, el estado funcional, y la etapa (I / II frente III / IV).
Debido a que la estatificación depende generalmente de la ubicación y del número de sitios afectados, no es una verdadera medida de carga tumoral, sin embargo, es un importante determinante para el pronóstico de LNH, y también para el programa de tratamiento utilizado.
Existe una puntuación general de pronóstico con valor en todas las variantes de LNH, como el Índice Pronóstico Internacional (IPI):
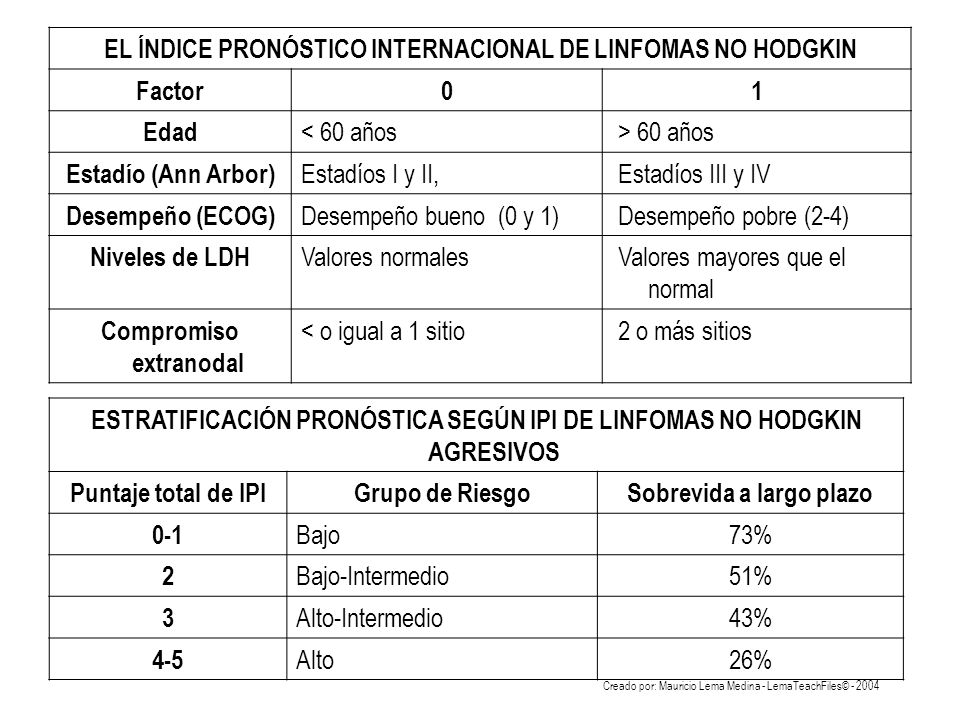
Existen diferentes estudios donde se analizaron a pacientes que presentaban LNH, y se separaron en grupos ya sea si estaban en tratamientos que incluían rituximab o no, de los que se extrapolaron diferentes tablas. A continuación, se detalla una tabla con puntajes de pronósticos ajustados por edad, donde las características mencionadas más arriba suman 1 punto cada una, exceptuando a la edad y el número de compromisos extranodales, que no se consideran. Obteniéndose un puntaje que varía desde 0 a 3.
|
PUNTAJE |
GRUPO DE RIESGO |
PORCENTAJE DE SUPERVIVENCIA (A 5 AÑOS) |
POCENTAJE DE REMISION COMPLETA |
|
0 |
Riesgo bajo |
56 |
91 |
|
1 |
Riesgo bajo o moderado |
44 |
71 |
|
2 |
Riesgo moderado o alto |
37 |
56 |
|
3 |
Riesgo alto |
21 |
36 |
9.- CASO CLÍNICO
Mujer de 68 años consulta en el policlínico de medicina, por haber notado aumento de volumen cervical, axilar e inguinal bilateral en forma progresiva, indoloros, en los últimos 7 meses. Notó además, sudoración nocturna, sin fiebre y baja de peso de 9 kilos en los últimos 5 meses. Antecedentes de Hipertensión arterial desde hace 4 años, bien controlada con enalapril. Al examen físico: buen estado nutritivo, palidez moderada de piel y mucosas, ganglios palpables de 4×3 cm en región cervical derecha, 2×2 cm en axila derecha e izquierda y varios de 2×1 cm en ambas regiones inguinales. Hígado en el reborde y bazo a 3 cm bajo el reborde costal. Extremidades: leve edema pierna izquierda.
Pregunta ¿Cuál es la conducta a seguir en esta paciente?
A) Hospitalizarla inmediatamente .
B) Enviarla a cirugía a tomar una biopsia ganglionar.
C) Controlarla en 3 meses más.
D) Indicar diuréticos, corticoides y control en 1 mes.
E) Indicar una transfusión de glóbulos rojos.
Respuesta correcta: B.
Comentario:
La conducta correcta es enviarla a tomar una biopsia ganglionar, ya que la primera posibilidad es un linfoma de tipo indolente. La biopsia es el examen más importante y el pronóstico depende del subtipo histológico. No se debe indicar corticoides, porque altera la histología, ya que los esteroides son linfolíticos. La transfusión de glóbulos rojos está indicada sólo si hay anemia sintomática. La paciente es ambulatoria, por lo tanto puede realizar el estudio en forma ambulatoria.
