
Este capítulo es tremendamente importante y además es bastante extenso, por lo que a continuación se intentará sintetizar los temas más relevantes, para que el medico no especialista conozca los conceptos más esenciales en la presentación clínica y manejo de estas patologías a su nivel y derivar adecuadamente según corresponda.
La piel es blanco de múltiples mecanismos de daño y en muchas enfermedades, los mecanismos autoinmunes son la explicación de la enfermedad y sus manifestaciones. Es así que la inmunología y la dermatología comparten campo de estudio.
Introducción
A nivel cutáneo el sistema inmune es bastante activo y complejo, tanto a nivel de inmunidad innata como en inmunidad adaptativa, tanto a nivel de dermis y epidermis. En la epidermis tenemos los queratinocitos y las células dendríticas (células de Langerhans en la piel) entre otros estirpes celulares.
A nivel de la dermis, para efectos de este capítulo, destacamos los macrófagos, los linfocitos T, linfocitos TCD8 y linfocitos TCD4, células natural killer, los mastocitos, que juegan un rol importantísimo en los mecanismos de enfermedad y respuesta cutánea.
En este capítulo estudiaremos tres grupos de patologías que tienen en común una fisiopatología preponderantemente inmunológica. Estas son las:
- Mesenquimopatías
- 1 Lupus
- 2 Dermatomiositis
- 3 Esclerodermia.
- Enfermedades ampollares.
- Mecanismos de hipersensibilidad.
- Urticaria
- Angioedema
- MESENQUIMOPATÍAS
Corresponden a un grupo extenso de enfermedades, que en general se estudian en reumatología, pero frecuentemente presentan compromiso cutáneo. Revisaremos lupus, dermatomiositis y esclerodermia. Es bueno recordar que frecuentemente que hay síndromes de sobreposición.
1.1 LUPUS ERITEMATOSO
El lupus corresponde a un espectro de enfermedades autoinmunes que afectan al tejido conectivo. Es multifactorial. Clínicamente la fotosensibilidad es característica.
De acuerdo a las manifestaciones cutáneas se pueden distinguir 3 entidades:
- Lupus eritematoso cutáneo crónico
- Lupus eritematoso cutáneo subagudo
- Lupus eritematoso sistémico
- LUPUS ERITEMATOSO CUTÁNEO CRÓNICO (LECC)
El lupus eritematoso cutáneo crónico el compromiso sistémico generalmente está ausente.
Es más frecuente en mujeres ( 2:1). Entre un 5 – 20% evolucionan a LES en distintas series. Se pueden observar anticuerpos antinucleares (ANA) >1:160 en un 5% de pacientes.
Existen varias formas clínicas: Lupus discoide, Lupus túmido, Paniculitis lúpica, lupus profundo, etc.
La forma clínica más frecuente del LECC es el lupus eritematoso discoide.
LUPUS ERITEMATOSO DISCOIDE .
Se presenta como pápulas y placas induradas, eritematosas, con escamas adherentes que puede evolucionar a cicatriz, atrofia, alteración de pigmentación y fibrosis.
Pueden haber clavos córneos, que son dilataciones de los orificios foliculares producto de la presencia de un tapón de queratina.
Puede ser localizado (60-80%) en el cual las lesiones de ubican sobre el cuello, comprometiendo con mayor frecuencia áreas fotoexpuestas como cara, cuero cabelludo y pabellones auriculares; y generalizado (20-40%), si las lesiones se ubican sobre y bajo el cuello . En este último caso las lesiones son más extensas, se asocian a mayor actividad clínica y con alteraciones de exámenes de laboratorio.
Con frecuencia este cuadro clínico se confunde con psoriasis, dermatitis de contacto o tiña, pero se pueden despejar dudas si la evolución es tórpida con el manejo básico de estas enfermedades o cronicidad del cuadro. La distribución en zonas fotoexpuestas y las cicatrices orientaría a LECC.
El lupus discoide tiende a exacerbarse con la exposición solar, apareciendo más lesiones
Si se piensa en Psoriasis y se intenta un raspado metódico de Brocq en un Lupus Discoide habría dolor y no se observarían las etapas características.
Se debe considerar que el lupus discoide, en fototipos oscuros generalmente dan hiperpigmentación postinflamatoria. Y cuando afecta cuero cabelludo, dejan alopecia cicatricial.
Manejo:
- Fotoprotección.
- Derivar.
Idealmente adjuntar estudio inicial incluyendo:
- ü Hemograma
- ü Perfil bioquímico
- ü Lipídico
- ü Hepático
- ü Orina completa y urocultivo
- ü Creatinina
- ü ANA, ENA y FR
El especialista podrá utilizar corticoides tópicos de alta potencia, como Clobetasol 0,05%, más algún antimalárico como hidroxicloroquina para el manejo.
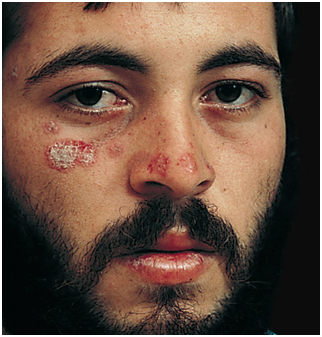
Figura 1: Lupus discoide (Fuente: http://bit.ly/2oG3ziE)
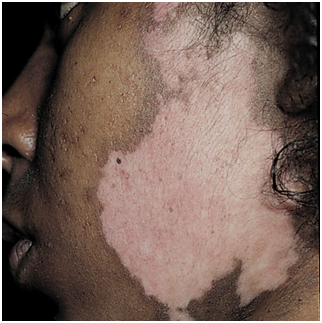
Figura 2: Lupus discoide atrófico (Fuente: http://bit.ly/2oG3ziE)
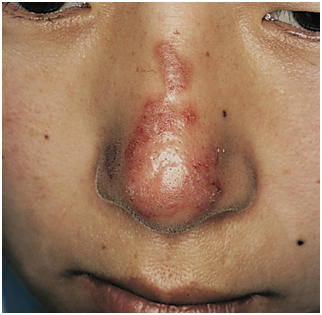
Figura 3: Lupus discoide en dorso nasal (Fuente: http://bit.ly/2oG3ziE)
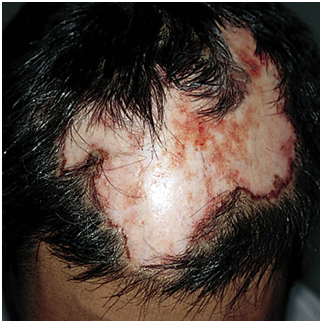
Figura 4: lupus discoide en cuero cabelludo, con alopecia cicatricial (Fuente: http://bit.ly/2oG3ziE)
- LUPUS ERITEMATOSO SUBAGUDO (LECSA)
El compromiso también se da en zonas fotoexpuestas, tiende a ser un poco más extenso afectando cara, cuello, tronco alto, superficie extensora de los brazos. Este tipo tiene una FOTOSENSIBILIDAD EXTREMA.
Este subtipo de lupus cutáneo no deja cicatrices ni atrofia sino más frecuentemente hipopigmentación residual. En LECSA hay mayor riesgo de compromiso sistémico. El 50% tiene al menos 4 criterios para LES, pero en general es menos severo que este. El paciente puede presentar artralgias, fiebre, malestar general y mialgias.
Entre un 10 a un 15% de los pacientes evolucionarán a LES en las distintas series.
Tiende a tener muchos anticuerpos positivos:
Anti Ro en el 70% de los pacientes
Anti La 25%
ANA mayor a 1:160 es positivo en 63% de los pacientes.
Las formas clínicas de LECSA:
- Anular policíclico
Lesiones anulares con un centro aclarado, con borde eritematoso y descamación.
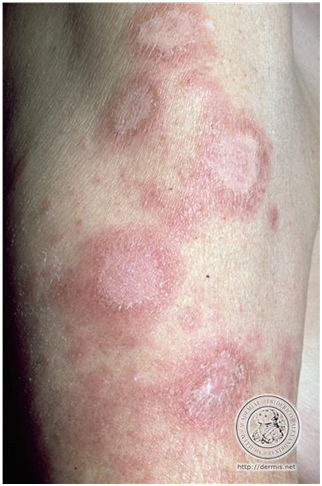
Figura 5: Lupus anular policíclico (Fuente: http://bit.ly/2pqU1sN)
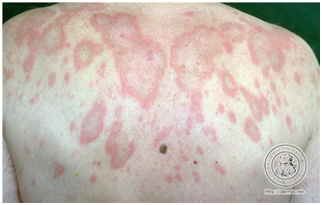
Figura 6: Lupus anular policíclico (Fuente: http://bit.ly/2pqU1sN)
- Psoriasiforme o papuloescamosa
Placas eritematoescamosas de borde bien definido parecido a las psoriasis.
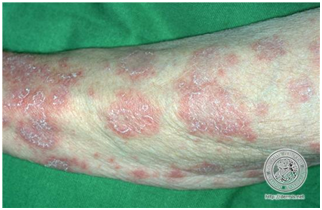
Figura 7: Lupus psoriasiforme (Fuente: http://bit.ly/2oEz8HG)
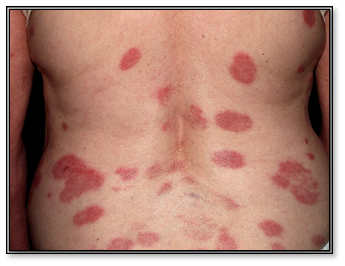
Figura 8: Lupus psoriasiforme (Fuente: http://bit.ly/2oED8bn)
Manejo:
- Fotoprotección.
- Derivar.
Idealmente adjuntar estudio inicial incluyendo:
- Hemograma
- Perfil bioquímico
- Lipídico
- Hepático
- Orina completa y urocultivo
- Creatinina
- ANA, ENA y FR
- LUPUS ERITEMATOSO SISTÉMICO
Es la enfermedad autoinmune clásica por excelencia. Se produce por la formación de autoanticuerpos y formación de complejos inmunes circulantes.
La proporción de mujeres/hombres es de 6:1.
Las manifestaciones clínicas cutáneas se ven en un 80-90% de los pacientes y por el compromiso sistémico, es una patología de manejo multidisciplinario.
Criterios clínicos e inmunológicos SLICC 2012
|
A. Criterios clínicos (al menos 1/11) |
|
1. Lupus cutáneo agudo: |
|
a. Rash malar Lupus buloso |
|
b. Necrólisis epidérmica tóxica del LES |
|
c. Rash maculopapular |
|
d. Rash lúpico fotosensible en ausencia de dermatomiositis |
|
e. Lupus cutáneo subagudo |
|
2. Lupus cutáneo crónico: |
|
a. Lupus discoide |
|
b. Lupus hipertrófico (verrugoso) |
|
c. Paniculitis lúpica |
|
d. Lupus en mucosas |
|
e. Lupus pernio |
|
f. Lupus túmido |
|
g. Sobreposición lupus discoide/liquen plano |
|
3. Úlceras orales (paladar, yugal, lengua) o nasales (en ausencia de otras causas como vasculitis, enfermedad de Behcet, infección (VHS), enfermedad inflamatoria intestinal, artritis reactiva) |
|
4. Alopecia no cicatricial (en ausencia de alopecia areata, drogas, déficit Fe y alopecia androgenética) |
|
5. Sinovitis (compromiso de 2 o más articulaciones dolorosas o con derrame o inflamadas y > 30 minutos de rigidez matinal) |
|
6. Serositis (en ausencia de otras causas como infección, uremia y pericarditis de Dressler). |
|
a. Pleuritis típica por >1 día o derrame pleural o frotes pleurales |
|
b. Dolor pericárdico típico > 1 día o derrame pericárdico o frote pericárdico o pericarditis (ECG) |
|
7. Compromiso renal: |
|
a. Relación proteinuria/creatininuria > o = 500 mg/24 hrs o |
|
b. Cilíndros hemáticos celulares |
|
8. Compromiso neurológico: |
|
a. Convulsiones |
|
b. Psicosis |
|
c. Mononeuritis múltiple (en ausencia de otras causas como vasculitis primaria) |
|
d. Mielitis |
|
e. Neuropatía periférica o craneal |
|
f. Estado confusional agudo (en ausencia de otras causas como tóxico/ metabólica, uremia, drogas |
|
9. Anemia hemolítica |
|
10. Leucopenia (<4.000/uL al menos en una oportunidad, en ausencia de otras causas como Sd. Felty, drogas o hipertensión portal) o linfopenia (<1.000/uL al menos en una oportunidad). |
|
11. Trombocitopenia (<100.000/uL al menos en una oportunidad, en ausencia de otras causas como drogas, hipertensión portal, púrpura trombótico trombocitopénico) |
|
B. Criterios inmunológicos (al menos 1/6): |
|
1. ANA+ |
|
2. Anti- dsDNA + |
|
3. Anti- Sm |
|
4. Ac antifosfolípidos: |
|
a. Anticoagulante lúpico- VDRL/RPR falso positivo |
|
b. Anticardiolipinas (IgA, IgG o IgM) |
|
c. Anti B2-glicoproteína I (IgA, IgG o IgM) |
|
5. Complemento bajo |
|
a. C3 bajo |
|
b. C4 bajo |
|
c. CH50 bajo |
|
6. Test de Coomb positivo (en ausencia de anemia hemolítica) |
A continuación se hará hincapié en las lesiones cutáneas del LES, que las podemos clasificar de manera didáctica en específicas e inespecíficas.
Manifestaciones específicas.
- LE agudo localizado, más conocido como ERITEMA MALAR.
Es la principal manifestación cutánea del LES, estando presente en el 50% de los pacientes.
Clínicamente se presenta como un eritema sobre las eminencias malares, el dorso nasal y que respeta el surco nasogeniano. Es una lesión cutánea específica de LES.
Evoluciona sin cicatriz.
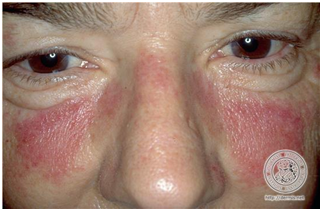
Figura 9: Eritema malar. “Mariposa lúpica” (Fuente: http://bit.ly/2pubv4T)
- Lupus eritematoso agudo generalizado.
Corresponde a un exantema maculopapular inespecífico, de área fotoexpuesta y palma de las manos, que es fotoexacerbada, y no evoluciona con cicatriz.
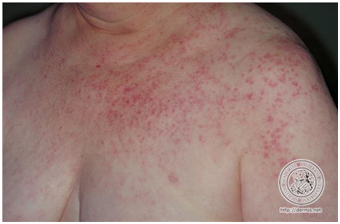
Figura 10: LEAG (Fuente: http://bit.ly/2oEM0xx)
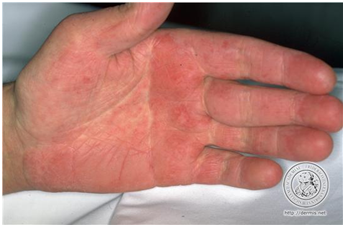
Figura 11: LEAG (Fuente: http://bit.ly/2oEM0xx)
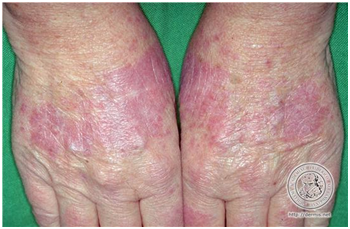
Figura 12: LEAG (Fuente: http://bit.ly/2oEM0xx)
Manifestaciones inespecíficas.
Alopecia difusa, fenómeno de Raynaud, lívedo reticularis, eritema palmar, hemorragias en astilla, proximales y marcadas.
Telangiectasias peringueales, Púrpura, Urticaria, Úlceras
Estas lesiones nunca están solas, y se presentan en el contexto de una mesenquimopatía.
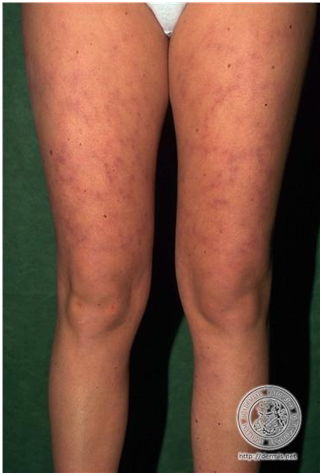
Figura 13: Lívedo reticularis (Fuente http://bit.ly/2oEM0xx)
Laboratorio en LES
En cuanto al laboratorio del LES, presenta muchos autoanticuerpos positivos.
- ANA:
- Es muy sensible (95%). Tiene un alto valor predictivo negativo ~ 100%
- Tiene baja especificidad (60%)
- Es sugerente de LES una dilución ≥ 1:160
- Anti DNA doble hebra:
- Es muy especifico para LES (95-99%), pero es poco sensible (30-40%). Es el mejor marcador de actividad de la enfermedad.
- ENA:
- Anti Sm: es el más especifico para LES (98-100%), poco sensible (8-40%)
- Anti-Ro: Sensibilidad: 32%
- Anti-La: Sensibilidad: 12%
- ü Hipocomplementemia: en períodos de actividad se observa C3 y C4 bajos.
- ü VDRL falso positivo (25%)
Manejo:
- Fotoprotección
- Derivar
Si las manifestaciones son de carácter grave ( ej: signos neurológicos, cardiacos, hepáticos y/o renales, etc) considerar la derivación a servicio de urgencia para hospitalización.
1.2. DERMATOMIOSITIS
Es una enfermedad inflamatoria de origen autoinmune: Esta miopatía inflamatoria proximal de músculos extensores se manifiesta como una debilidad proximal, asociada a erupción cutánea característica. Se ve con más frecuencia en mujeres que en hombres en una razón 2:1. Tiene 2 peaks de incidencia, uno infantil en menores de 10 años y en adultos entre los 45-60 años.
Existen variantes sin lesiones en piel (polimiositis) y otras solo con lesiones en piel sin compromiso muscular (dermatomiositis amiopática)
Se considera un síndrome para neoplásico dado que puede asociarse a adenocarcinomas ( ovaricos, pulmón, gastrointestinal) antes, durante y despues.
Clínica:
En un 40% de pacientes el compromiso cutáneo es la primera manifestación y es extremadamente fotosensible y muy pruriginosa.
El compromiso muscular se puede ver antes, durante o años después de las manifestaciones cutáneas.
Manifestación cutánea:
- Eritema heliotropo
Es un Rash eritematovioláceo en zona periorbitaria y párpados. Puede asociar o no edema. Está presente entre el 30 y 60% de los casos.
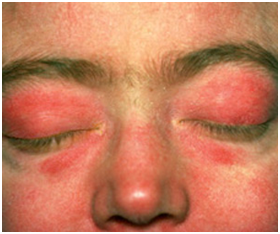
Figura 14: Rash heliotropo (Fuente: http://bit.ly/2pJOdau)
- Pápulas de Gottron
Placas o pápulas eritematovioláceas sobre prominencias óseas en articulaciones MCF, IF, codos, rodillas o tobillos. Es clásicamente considerado un signo patognomónico de la enfermedad y se puede ver en hasta el 70% de los pacientes.
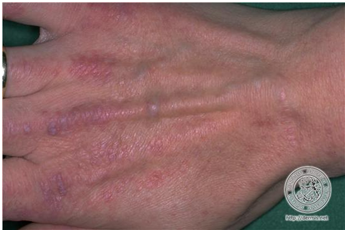
Figura 15: Pápulas de Gottron (Fuente: http://bit.ly/2oG83pi)
- Signo de Gottron
Signo de Gottron se refiere a cambio de coloración violácea de nudillos, codos y rodillas. No son pápulas.
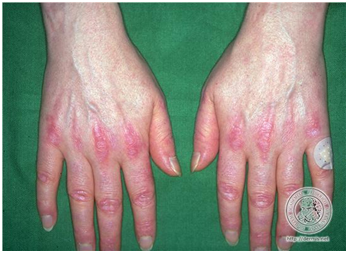
Figura 16: Signo de Gottron (Fuente: http://bit.ly/2oG83pi)
- Poiquilodermia
Corresponde a la combinación de atrofia , telangiectasias, hipo o híper pigmentación en una misma lesión. Suele verse en cara, cuello, tronco y EESS.
Cuando compromete la zona de V cuello y espalda se habla del signo del chal.
No es específico de la enfermedad.
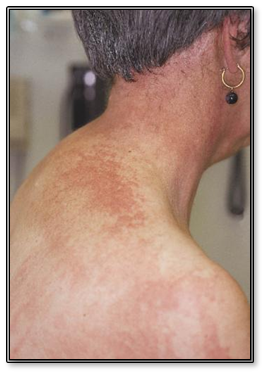
Figura 17: Poiquilodermia (Fuente: http://bit.ly/2pK7M2k)
- Signo de la pistolera
Recibe el nombre por su ubicación. Son máculas eritemato-violáceas en las caras laterales de muslos y cadera, y son simétricas.
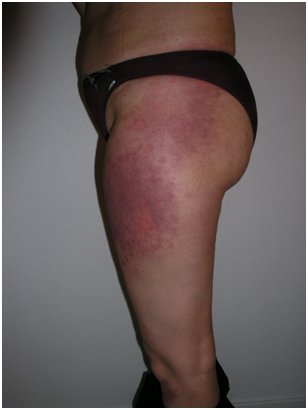
Figura 18: Signo de la pistolera (Fuente: http://bit.ly/2paD6eb)
Además de lo descrito, pueden haber otras manifestaciones cutáneas como las telangiectasias periungueales (que también se ven en lupus), asociado a eritema periungueal. Hay distrofia de la cutícula, se engruesa. Finalmente se ven infartos hemorrágicos peringueales.
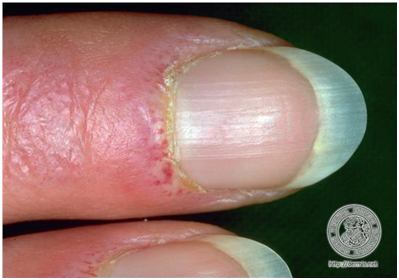
Figura 19: Manifestaciones periungueales de DM. (Fuente: http://bit.ly/2oG83pi)
Clínica muscular:
Debilidad muscular bilateral proximal, simétrica
Cintura escapular y pelviana: Dificultad para peinarse, levantar la cabeza, pararse
Otros:
Artralgias, artritis no erosiva, disnea (debilidad músculos respiratorios, fibrosis intersticial, mialgias, fatiga, arritmias por miocarditis, disfonía, disfagia.
Criterios diagnósticos de Dermatomiositis
- Debilidad muscular simétrica, proximal, progresiva.
- Biopsia muscular con miopatía inflamatoria.
- Aumento niveles séricos enzimas musculares.
- Hallazgos EMG de miopatía.
- Erupción cutánea típica de DM* (criterio necesario para diagnóstico DM)
4 criterios presentes: Dg. definitivo
3 criterios presentes: Dg. probable
2 criterios presentes: Dg. posible
Manejo:
- Fotoprotección.
- Derivar.
Idealmente adjuntar estudio inicial:
- Hemograma
- CK total
- Perfil bioquímico, lipídico, hepático.
- Orina completa y urocultivo
- Creatinina y ANA, si se puede ENA y FR).
- Fotoprotección.
Si las manifestaciones son de carácter grave ( ej: signos neurológicos , cardiacos, hepáticos y/o renales, etc) considerar la derivación a servicio de urgencia para hospitalización.
1.3 ESCLERODERMIA
Al igual que el lupus, la esclerodermia es un espectro de enfermedades en que se produce aumento del depósito de colágeno a nivel de la dermis y que puede ser localizada o sistémica.
La forma localizada se conoce como morfea, corresponde a la presentación más frecuente y compromete solo la piel y subcutáneo, sin comprometer el resto de los órganos.
La forma sistémica se llama esclerosis sistémica progresiva o esclerodermia (como sinónimos) compromete la piel, subcutáneo y los órganos internos.
MORFEA
Es una esclerodermia circunscrita y limitada a la piel. No presenta fenómeno de Raynaud y es generalmente autolimitada. Cuando se asocia morfea y fenómeno de Raynaud, probablemente evolucione a una esclerodermia. Tiene varias formas de presentación clínica dentro de las que destaca:
- En Placas
- En Gotas
- Lineal
- – En Golpe de sable
- Generalizada
- Profunda
- Morfea en placas
Es la forma más frecuente en adultos.
Se presenta como múltiples placas, asimétricas, violáceas, induradas que luego pierden su color en la parte central y adquiere un centro esclerótico, cicatricial, color blanco brillante, borde indurado violáceo (que indica actividad. Además asocia pérdida de anexos cutáneos, menor sudoración, hipoestesia. Deja una hiperpigmentación residual. Progresa por 3-5 años, luego se detiene.
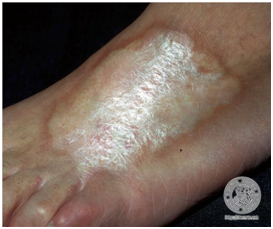
Figura 20: Morfea en placa (Fuente: http://bit.ly/2paFpy3)
- Morfea lineal
Es la forma más frecuente en niños, compromete las extremidades o cara. En general es unilateral, puede tener compromiso óseo, músculo, tendones.
En extremidades si es muy profunda puede dejar bandas cicatricial que compromete movilidad y crecimiento extremidad con graves secuelas.
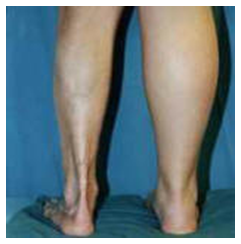
Figura 21: Morfea lineal (Fuente: http://bit.ly/2paFpy3)
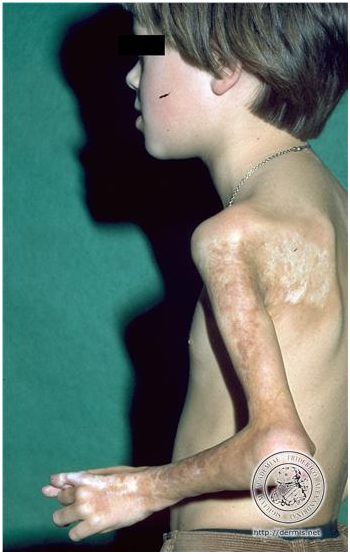
Figura 22: Bandas cicatriciales (fuente: http://bit.ly/2paFpy3)
2.1 Morfea en golpe de sable
Es una variedad de morfea lineal, que es paramedial, en región frontoparietal. Es muy profunda y se asocia a eritema y edema indurado, luego atrofia facial y esclerosis. Puede dar compromiso óseo y del SNC.
En casos severos puede llevar a una forma severa de hemiatrofia facial, conocida como síndrome de Parry-Romberg
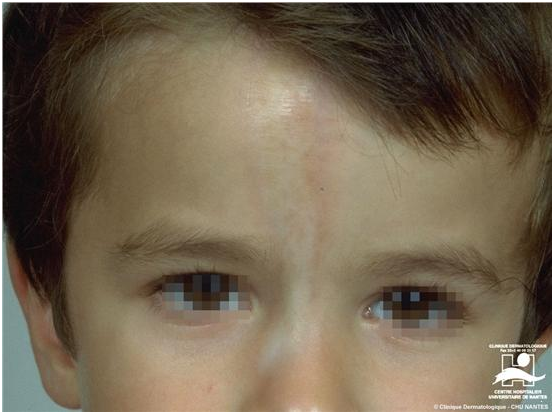
Figura 22: Morfea lineal en golpe de sable (Fuente: http://bit.ly/2paFpy3)
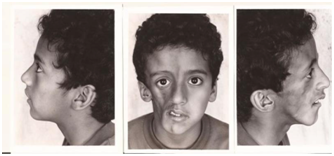
Figura 23: hemiatrofia facial (Síndrome Parry-Romberg) (Fuente: http://bit.ly/2oiqEFd)
- Morfea generalizada
Igual a la morfea en placas, pero estas confluyen hasta compromiso generalizado, que puede llevar a contracturas incapacitantes, pero no se asocia a esclerosis sistémica.
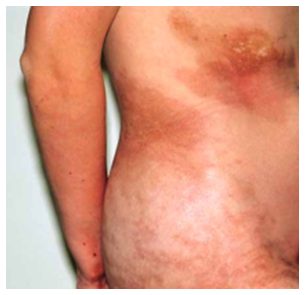
Figura 24: morfea generalizada (Fuente: http://bit.ly/2oEJesc)
Manejo: Fotoprotección y derivar con el estudio
ESCLERODERMIA
A diferencia de la morfea, presenta:
- Fenómeno de Raynaud, en el 90% de los pacientes. Generalmente precede a esclerosis. Es un trastorno paroxístico vasoespástico de los dedos gatillado por frío, que clínicamente se presenta por cambio de coloración de estos. Una primera fase de vasoconstricción torna los dedos de color blanco,
luego azul y cuando se reperfunde completamente, intensamente rojos. No duele pero sí molesta. Los casos severos pueden dar úlceras. No confundir con el eritema pernio o sabañones, que es un eritema violáceo de los dedos, muy pruriginoso y levemente doloroso.
La fase blanca es la que debe estar para el diagnóstico.
- Induración simétrica y difusa de la piel
Tiene 3 fases: una edematosa, una indurativa y una atrofica.
- ü Edematosa: piel inflamada, eritematosa y pruriginosa. Simétrica en mano. Apariencia de la piel “sal y pimienta”, debido a zonas de hipo e hiperpigmentación.
- ü Indurativa: endurecimiento y engrosamiento (apariencia lustrosa y sin pliegues pliegues. Dura 2-3 años.
- ü Atrofica: piel tensa sobre areas de flexion-ulceras dolorosas.
Esclerodactilia, engrosamiento y endurecimiento de los dedos
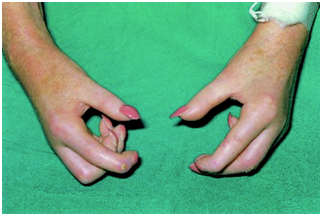
Figura 25: Esclerodactilia (Fuente: http://bit.ly/2oiA1EO)
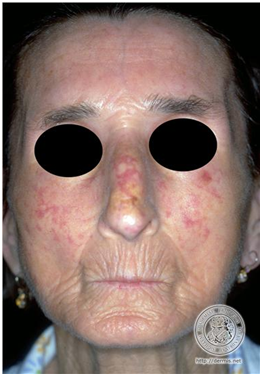
Figura 26: Facie esclerodérmica (Fuente: http://bit.ly/2pupQ1c).
- Compromiso sistémico
Tiene una forma limitada y difusa.
La forma limitada era lo que se conocía como síndrome de CREST, concepto que en la actualidad no se utiliza mucho, que era acrónimo para: calcinosis, Raynaud, esofagopatía, esclerodactilia y telangiectasias). En la variedad limitada, la piel puede presentar calcinosis subcutánea, que corresponde al depósito cutáneo de hidroxiapatita, presenta capilaroscopía alterada, dado capilares distorsionados y amputados. Se ven además telangiectasias.
Cuando tiene compromiso facial se puede ver disminución de la apertura oral y ocular, labios finos y rígidos, frenillo lingual nacarado y reabsorción ósea de la mandíbula.
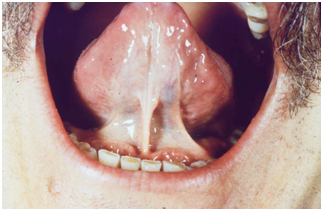
Figura 27: Frenillo lingual nacarado (Fuente: http://bit.ly/2pK15gx)
La forma sistémica tiene compromiso:
- Pulmonar: que es la mayor causa de mortalidad actualmente.
- Cardiaco: repercusión de la Hipertensión pulmonar
- Digestivo: más frecuente y precoz. Da esofagopatía con disfagia.
- Renal: como HTA y falla renal rápidamente progresiva.
- Otros: síndrome de Sicca, patología dental severa, neuropatías por atrapamiento, hipotiroidismo, colangitis biliar primaria, hepatitis autoinmune, disfunción sexual.
Laboratorio
La variedad limitada: presenta ANA positivo, patrón anticentrómero. Presenta mayor riesgo de hipertensión pulmonar. Tiene mejor pronóstico que la variedad difusa.
La variedad difusa: tiene compromiso visceral significativo. Tiene ANA positivo, y destaca ENA Scl-70. Tiene mayor riesgo de fibrosis pulmonar. Tiene mal pronóstico.
Manejo: Fotoprotección y derivar con el laboratorio ad hoc.
- ENFERMEDADES AMPOLLARES
Corresponden a un grupo de enfermedades mucocutáneas, caracterizadas por la formación de ampollas o bulas. Tiene diferentes etiologías: infecciosa, medicamentosa, autoinmune. Solo estudiaremos las autoinmunes en este capítulo.
Enfermedades ampollares autoinmunes
Grupo de enfermedades que presentan anticuerpos dirigidos a proteínas que forman parte de las uniones intercelulares.
|
Uniones intercelulares intraepidérmicas (Desmosomas y/o Uniones adherentes): |
|
PÉNFIGOS |
|
1. Pénfigo foliáceo |
|
2. Pénfigo eritematoso (Sanear Usher) |
|
3. Pénfigo vulgar |
|
4. Pénfigo vegetante |
|
5. Pénfigo IgA |
|
6. Pénfigo paraneoplásico |
|
7. Pénfigo inducido por drogas |
Una ampolla formada al interior de la epidermis, es producto de la acción de anticuerpos anti proteínas de unión de queratinocitos.
|
Uniones dermo-epidérmicas (Hemidesmosomas y Afectación de Membrana basal): |
|
PENFIGOIDES |
|
1. Penfigoide ampollar |
|
2. Penfigoide cicatricial |
|
3. Dermatosis lineal IgA |
|
4. Penfigoide del embarazo |
|
5. Epidermolisis bulosa adquirida |
|
6. Lupus eritematoso buloso |
|
7. Dermatitis herpetiforme |
La ampolla formada a nivel subepidérmico, es producto de la acción de anticuerpos anti proteínas de la unión dermoepidérmica.
Dado que las enfermedades ampollares son muchas, en este capítulo solo se revisaran el pénfigo vulgar, vegetante y foliáceo; el penfigoide buloso, herpes gestationalis y cicatricial y la dermatitis herpetiforme.
PÉNFIGO
Son enfermedades autoinmunes caracterizadas por anticuerpos anti proteínas que forman las uniones entre queratinocitos. Se caracterizan por:
- Acantolisis, que es la ruptura y fragmentación de los desmosomas, con pérdida de la cohesión entre las células, que resulta en vesícula o bula.
- La ampolla es intraepidérmica, flácida, que casi nunca se ve. Es frecuente ver erosiones, porque el techo de la ampolla es muy delgado
- Tiene signo Nikolsky positivo
- Olor fétido sui generis
Los niveles de autoanticuerpos séricos se correlacionan con el nivel de actividad de la enfermedad, que no ocurren en otras enfermedades ampollares autoinmunes, son predominantemente de tipo IgG por lo que atraviesan la placenta y el paso de autoanticuerpos al RN puede generar una enfermedad ampollar transitoria
Signo de Nikolsky.
El signo de Nikolsky es un indicador de una acantolisis activa.
Es el desprendimiento epidérmico provocado por un movimiento de presión realizado con el dedo sobre la piel sana perilesional.
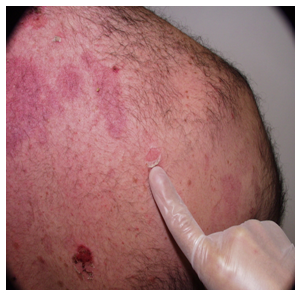
Figura 28: Signo de Nikolsky (Fuente: Propia)
Los autoanticuerpos que se depositan más frecuentemente son IgG, complemento, IgM o IgA.
Pénfigo vulgar
Es la forma más frecuente. Corresponde al 80% de los pénfigos, aparece entre los 30 y 60 años, sin preferencia de sexo. Evoluciona en brotes.
La ampolla deja erosiones que epiteliza sin dejar cicatriz. Bajo la ampolla queda un tejido sangrante.
Se observa compromiso mucoso en 60% de los pacientes.
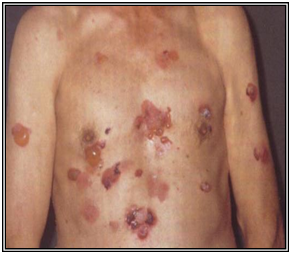
Figura 29 : Pénfigo vulgar (Fuente: http://bit.ly/2oETSz5)
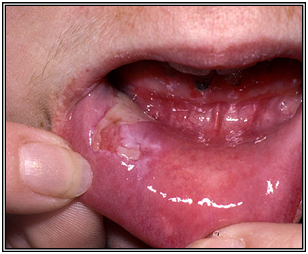
Figura 30: Compromiso de mucosa oral en pénfigo vulgar (Fuente: http://bit.ly/2ohZkXW)
Pénfigo vegetante
Se forman lesiones verrucosas asociadas a vesículas, desde el inicio del cuadro, en zonas intertriginosas, cuero cabelludo y cara. Por lo general las formas verrucosas aparecen sobre piel sana o erosionada.
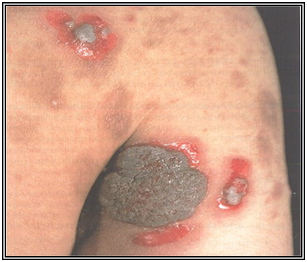
Figura 31: Pénfigo vegetante (Fuente: Propia)
Pénfigo foliáceo
La ampolla es subcórnea, predomina en zonas más seborreicas. No afecta mucosas y tiene variedades epidémicas. En Brasil hay una variedad transmitida por mosquito (Fogo Salvagem)
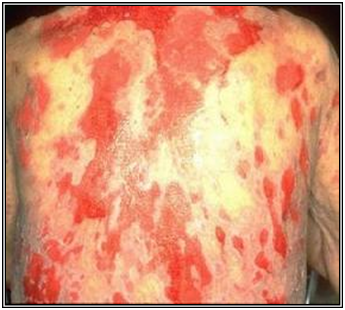
Figura 32: Pénfigo foliáceo (Fuente: http://bit.ly/2onhFlB)
PENFIGOIDE
Son enfermedades autoinmunes caracterizadas por autoanticuerpos tipo IgG o IgA anti-membrana basal o contra proteínas que forman parte de la unión dermoepidermica. La ampolla es subepidermica, por lo que el signo de Nikolsky será negativo.
Los niveles de autoanticuerpos séricos no se correlacionan con el nivel de actividad de la enfermedad, que lo diferencia de los pénfigos.
Penfigoide buloso
Es el más frecuente de TODAS las enfermedades ampollares.
Se presenta en adulto mayor 65 a 70 años. No tiene preferencias de sexo o raza.
Clínica:
Presenta una fase prodrómica inespecífica, pruriginosa. Parte como una reacción urticarial inespecífica, que evoluciona a ampollas tensas sobre base eritematosa o piel normal. Al romperse evolucionan a la reepitelización y dejan lesiones residuales pigmentarias que duran mucho tiempo.
Las zonas más frecuentemente comprometidas son los pliegues, superficie flexora EEs, pliegues, abdomen. Las lesiones en mucosas es raro y más leve, generalmente en la boca.
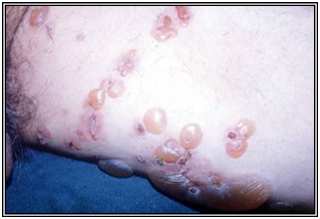
Figura 33: Penfigoide buloso (Fuente: Propia)
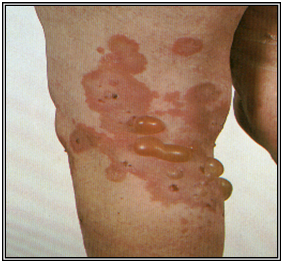
Figura 34: Penfigoide buloso (Fuente: Propia)
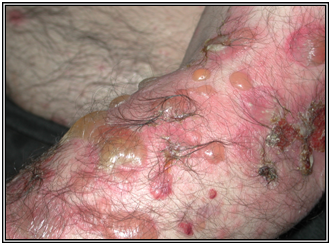
Figura 35: Penfigoide buloso (Fuente: Propia)
Dermatosis por IgA lineal
Presenta dos formas clínicas:
- Infantil: aparece después de los 2 años desaparece en la pubertad
- Adulto: se presenta en adultos jóvenes, se presenta como un penfigoide polimorfo). Lo característico son ampollas ovaladas con ampollas alrededor, que dan un aspecto de roseta. Tiene compromiso de mucosas 1/3 de los pacientes.
Penfigoide del embarazo (Herpes gestationis)
Dermatosis ampollar específica que ocurre sólo en el embarazo. Ocurre en el segundo y tercer trimestre. Menos en post parto.
Clínica:
Prurito en abdomen, luego erupción urticarial sobre piel eritematosas o normal. No afecta mucosas. Cede tras el parto o puerperio.
Los RN tienen lesiones en un 10%. No es grave, no se asocia a muerte fetal.
Puede recurrir en distintos embarazos.
Penfigoide cicatricial
Se da en ancianos.
Clínicamente se presenta con erosiones y lesiones en mucosas periorificiales, ojos y boca.
No afecta la piel.
Puede dar gingivoestomatitis erosiva más frecuentemente, luego compromiso conjuntival.
Las cicatrices que se forman, y esto es lo grave, dejan sinequias en boca y parpados.
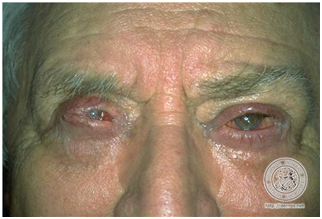
Figura 36: Penfigoide cicatricial (Fuente: http://bit.ly/2oELyPQ)
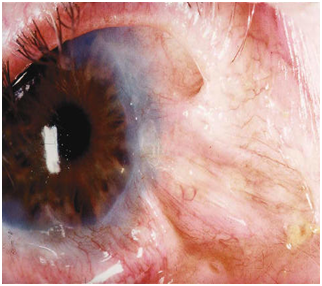
Figura 37: Sinequias palpebrales (Fuente: http://bit.ly/2nVKM44)
DERMATITIS HERPETIFORME
Pertenece al grupo de enfermedades ampollares autoinmunes subepidérmicas.
Aparece entre los 25 a 55 años, más típicamente en mujeres. El 85-90% está asociado a HLA-B8 y DRw3, esto la hace diferente al resto de las enfermedades ampollares.
Clínica:
Se presenta como una erupción pápulo vesicular agrupada (“patrón herpetiforme”) intensamente pruriginosas. No tiene nada que ver con la infección por virus herpes.
Se ve en superficie extensora de extremidades y glúteos. Compromiso de nuca, escapula, glúteos, muslos atrás, rodillas, codos antebrazos, lumbar.
Tiene disposición simétrica, con escaso compromiso mucoso.
Está asociada a enteropatía por gluten (Enfermedad Celiaca).
El tratamiento de elección es la dapsona y dieta libre de gluten.
MANEJO DE ENFERMEDADES AMPOLLARES
Derivar para estudio y manejo. El estudio es con biopsia e inmunofluorescencia directa.
Importante que en los casos extensos y con compromiso del estado general derivar urgente para manejo de soporte hospitalizado.
Previo a derivar manejar el prurito con antihistamínicos y cubrir con apósito estériles las zonas denudadas.
- MECANISMOS DE HIPERSENSIBILIDAD
Situaciones en las que se produce una respuesta inmunitaria adaptativa exagerada o inadecuada.
La hipersensibilidad no se manifiesta tras la primera exposición al antígeno (Ag), sino que suele aparecer tras un contacto posterior.
Los mecanismos se clasifican en 4 tipos, según Gell y Coombs.
Tipo 1: Mediada por IgE
Mediada por mastocitos, que producen IgE.
Son el mecanismo que explica el asma, la urticaria aguda, la rinitis alérgica y el shock anafiláctico.
Tipo 2: Citotoxicidad dependiente de anticuerpos.
Se producen anticuerpos (salvo IgE) contra antígenos. Unión antígeno anticuerpo, en la superficie celular. Esta interacción activa fagocitos y lisis mediada por complemento.
Es el mecanismo de algunas alergias a drogas, alopecia areata, vitíligo, liquen plano.
Tipo 3: Complejos inmunes circulantes
Mecanismo de daño caracterizado por unión antígeno anticuerpo, formación de complejo inmune circulante. Estos se depositan en los tejidos, activan la vía del complemento y atraen PMN. Se producen lesiones tisulares e inflamación.
Es el mecanismo de urticaria vasculítica, glomerulonefritis.
Tipo 4: Inmunidad mediada por linfocitos T sensibilizados.
Mecanismo de daño producido por la activación de células inmune, especialmente linfocitos T. Reacción retardada, hipersensibilidad tardía, respuesta celular. Hay 3 subtipos de HS IV.
Explica el mecanismo de la dermatitis de contacto. .
Ejemplo: Sarcoidosis.
A continuación estudiaremos la urticaria, como cuadro clínico característico del mecanismo de hipersensibilidad tipo 1.
URTICARIA
Es una reacción inflamatoria de la piel con eritema, vasodilatación, pseudopápulas habones. Se define como aguda un cuadro de menos de 6 semanas y crónica cuando esta sobrepasa las 6 semanas.
Epidemiología
El 15 -20% de la población puede llegar a presentar un cuadro de urticaria aguda durante su vida.
La urticaria crónica puede estar presente en el 25% de las personas que han tenido urticaria aguda.
Mecanismo: responde a un mecanismo de hipersensibilidad tipo 1.
El antígeno se une a una IgE específica que se une al mastocito induciendo su degranulación y liberación de mediadores.
Sin embargo la degranulación del mastocito puede ser por mecanismos inmunológicos y no inmunológicos.
- Dentro de los inmunológicos: mediados por IgE, autoanticuerpos contra receptor de IgE, complejos inmunes.
- Dentro de los no inmunológicos: acción directa sobre mastocito como medicamentos, aines, mastocitosis, actividad de complemento, infecciones parasitarias.
Diagnóstico
La historia clínica es suficiente para hacer diagnóstico de urticaria aguda. En general no dura más de 4 a 7 días. Se habla de urticaria crónica cuando dura más de 6 semanas con periodos con habones alternantes con piel sana.
Los habones pruriginosos aparecen en cualquier punto de la superficie cutánea, característicamente una misma lesión no suele durar más de 24 horas. NO PUEDE durar más de 24 horas. Si dura más de 24 horas, reconsiderar el diagnóstico ( es un habón?) Por otro lado una habón de más de 24 horas sin cambio alguno nos debe hacer sospechar en urticaria vasculítica, cuadro que , como dice su nombre , es una vasculitis y se puede asociar a otras enfermedades del mesénquima.
También estos cuadros se pueden acompañar a angioedema ( tumefacción dérmica profunda localizada y/o hipodermis asociado a aumento de la permeabilidad vascular) .
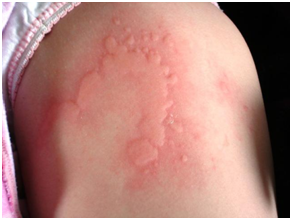
Figura 38: Habones o pseudopápulas (Fuente: http://bit.ly/2puo2VO)
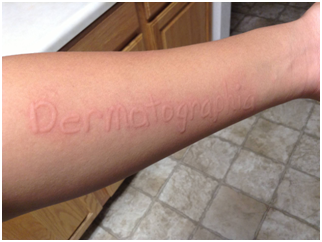
Figura 39: Dermografismo (Fuente: http://bit.ly/2pr5oRK).
Estudio:
En Urticaria Aguda NO amerita estudio. Las causas son difíciles de reconocer. Hasta en 50% de los casos de desconoce la causa, puede asociarse a infecciones respiratorias superiores , intolerancia alimentarias, fármacos , etc.
Frente a una urticaria crónica o múltiples episodios de urticaria aguda o angioedema se justifica el derivar. Se sugiere solicitar un chequeo general de laboratorio al momento de derivar :
Exámenes generales incluyen: Hemograma, VHS, PCR, Perfil bioquímico y orina completa con urocultivo.
Manejo urticaria aguda.
- – Evitar contacto con agente sospechoso (medicamento, alimentos ricos en histamina)
- – Evitar gatillantes físicos de urticaria ( con el fin de no empeorar los síntomas: calor, alimentos ricos de histamina etc)
- – Tratamiento con antihistamínicos H1: Son la primera línea. Se puede usar cualquiera en dosis habituales.
- – En caso de poca respuesta o asociacion a angioedema de puede usar: Prednisona 0,5 a 1 mg/kg dia, por 7 días.
Manejo urticaria aguda.
Debe derivarse a especialista para estudio. Se puede iniciar antihistamínicos H1 de tercera generación (levocetirizina, la fexofenadina, desloratadina). Estos fármacos se pueden aumentar hasta 4 veces su dosis habitual si no mejoran los síntomas durante el manejo especializado. Aun hay disparidad de criterios en como manejar estos cuadros cuando no responden a la primera línea.
Signos de alarma en Urticaria aguda y Anafilaxia
- Prurito intenso de manos y nuca.
- Angioedema labios, lengua
- Dificultad respiratoria
La presencia de cualquiera de estos nos debe hacer sospechar en una anafilaxia (dos sistemas comprometidos) o un shock anafiláctico, que es una emergencia médica por la obstrucción vía aérea por edema laríngeo.
El tratamiento debe ser inmediato con adrenalina (evita la obstrucción vía aérea) y luego corticoides ( pensando en la segunda fase de la activación de mastocitos con liberación de citoquinas y el antihistamínico pensando en los síntomas de la urticaria).
Medidas generales:
Levantar piernas, oxígeno
Medidas específicas:
Adrenalina:
En adultos:
Adrenalina 0,3-0,5 mg intramuscular (1:1.000)
En niños:
Adrenalina 0,01 mg/kg (dosis máxima 0,3 mg/kg)
Corticoide:
Hidrocortisona 100-300 mg en adultos;
Hidrocortisona 25 mg en <1 año.
Hidrocortisona 50 mg de 1-5 años.
Hidrocortisona 100 mg de 6 a 12 años ev en infusión lenta.
Antihistamínico
Clorfenamina 10-20 mg día ev o 0,2-0,4 mg/kg/día en niños.
