Generalidades
Se denominan síndromes mieloproliferativos crónicos (SMPc) a aquellas panmielopatías clonales, en las que la mutación de la célula germinal pluripotencial tiene como característica la proliferación excesiva, dando lugar a un incremento de las series hematopoyéticas. El incremento de las series hematopoyéticas se da tanto en la sangre periférica como en la médula ósea, pero habitualmente existe predominio en alguna de las series.
Se reconocen 4 SMC, que se distinguen principalmente por la serie en la que existe predominio:
- Policitemia vera: predomina la serie roja.
- Leucemia mieloide crónica: predomina la serie blanca.
- Trombocitemia esencial: predomina la serie megacariocítica-plaquetaria.
- Mielofibrosis primaria con metaplasia mieloide o mielofibrosis agnogénica: predominio de proceso formador de tejido fibroso colagénico.

Policitemia Vera (PV):
La policitemia vera es una neoplasia mieloproliferativa crónica caracterizada por un aumento de la producción de las células rojas, independientemente de los mecanismos que regulan la eritropoyesis normal, es decir, independiente de los niveles de eritropoyetina (EPO).
De esta manera, la diferencia fundamental con el resto de las situaciones en las que incrementan los glóbulos rojos (poliglobulias secundarias) es que en estas últimas existe un exceso de EPO.
Más del 95% de los casos de PV son portadores de la mutación JAK2V617F de la tirosina-cinasa JAK2 en el cromosoma 9p. Dicha mutación permite un incremento en la proliferación y diferenciación celular hematopoyética e inhibe la apoptsis , con formación espontánea de colonias eritroides.
Se trata de un trastorno que aparece habitualmente en varones de edad media y de causa desconocida, con una incidencia anual de 1-3 casos/100.000.
Clínica
Fase proliferativa o de plétora eritrocitaria: se caracteriza por signos y síntomas tales como la rubicundez cutánea o mucosa, cefaleas, acúfenos, mareos, parestesias, trastornos neurológicos derivados de la dificultad de la circulación sanguínea en el cerebro, hiperviscosidad, trombosis, hemorragias (por alteración de la función plaquetaria por hiperviscosidad), síntomas de hipermetabolismo tales como la disminución de peso y la sudoración nocturna, hipersensibilidad ósea, prurito (por incremento de la secreción de histamina al incrementarse la cantidad de basófilos) e hipertensión arterial por el aumento de la viscosidad de la sangre.
Como características propias de todos los síndromes mieloproliferativos, la policitemia vera suele cursar con esplenomegalia en el 75% de los casos y hepatomegalia, hasta en la tercera parte.
Como anomalías de laboratorio, encontraremos:
– Incremento del número de hematíes, disminución del volumen corpuscular medio (por disminución del hierro en cada hematíe).
– Respecto a la serie blanca, existe un incremento de leucocitos, fundamentalmente neutrófilos, que, a diferencia de la leucemia mieloide crónica o la hemoglobinuria paroxística nocturna, presentan un aumento de la fosfatasa alcalina leucocitaria.
– Existe una disminución de la eritropoyetina sérica y un aumento de la vitamina B12 sérica (por el aumento de las transcobalaminas I y III derivadas de los neutrófilos).
– Hay también trombocitosis con alteración del funcionamiento plaquetario.
– En la médula ósea nos encontraremos una hiperplasia de las tres series, predominantemente de la serie roja.
– Detección de JAK2V817F u otras mutaciones similares (JAK2 Exon 12)
Fase de metamorfosis: se produce una disminución progresiva de la proliferación clonal, lo que se traduce en una disminución progresiva del hematocrito, que puede incluso ocasionar anemia, y una tendencia progresiva a fibrosis medular. A esta fase se la conoce también con el nombre de policitemia vera gastada. La transformación puede ocurrir de varios meses a varios años después del diagnóstico de policitemia vera.
Diagnóstico:
Se hace el diagnóstico de PV cuando se cumplen los 3 criterios mayores o los dos primeros criterio mayores y dos criterio menores.
Criterios mayores:
– Aumento de la masa eritrocitaria (en varones 36 ml/Kg y en mujeres 32 ml/Kg)
– Saturación arterial de oxígeno 92%
– Esplenomegalia
Criterios menores:
– Trombocitosis superior a 400.000 por mm3
– Leucocitosis superior a 12.000 por mm3 en ausencia de fiebre o infección
– Fosfatasa alcalina leucocitaria superior a 100 en ausencia de fiebre o infección.
– Vitamina B12 sérica superior a 900 picogramos por ml
Tratamiento
– Pacientes con PV de bajo riesgo (menores de 60 años, sin eventos CV previos): sangrías, con el fin de alcanzar un hematocrito menor del 45% en hombres y menor del 47% en mujeres.
– Pacientes con PV de riesgo alto (mayores de 60 con eventos CV previos): tratamiento mielosupresor con hidroxiurea (en caso de pacientes menores de 50 años y mujeres en edad fértil se recomienda mielosuprimir con interferón ) Otros fármacos empleados en PV pueden ser AAS y anagrelida si la trombocitosis es muy severa (disminuye la producción megacariocítica).
Pronóstico:
Sin tratamiento, la vida media de los pacientes con policitemia vera es de un año y medio, con tratamiento, pueden llegar a vivir hasta 10 años. Como todos los procesos de la célula germinal pluripotencial, se trata de una enfermedad incurable, a menos que se realice un trasplante de medula ósea.
La causa más frecuente de muerte en la policitemia vera son las trombosis (hasta un tercio de los casos de muerte son provocados por trombosis).
Mielofibrosis primaria (MP):
Se trata de una panmielopatía clonal que se acompaña de mielofibrosis reactiva, hematopoyesis extramedular (metaplasia mieloide) y mieloptisis.
El evento inicial en la enfermedad consiste en una proliferación de megacariocitos en la médula ósea, con su muerte intramedular y liberación local de varios factores estimuladores de los fibroblastos, y por tanto de la formación de tejido fibroso, apareciendo la fibrosis medular.
Cuando la fibrosis medular es extensa, se produce la emigración de las células germinales pluripotenciales de la médula ósea, produciendo hematopoyesis extramedular principalmente en el bazo y en el hígado. Encontraremos esplenomegalia en casi todos los casos y hepatomegalia hasta en el 50%.
Dado que la enfermedad se caracteriza por una fibrosis medular y mieloptisis (ocupación de la médula normal), es característica la presencia en sangre periférica de la denominada reacción leucoeritroblástica (formas jóvenes de todas las series hematopoyéticas) y dacriocitos o hematíes en forma de lágrima.
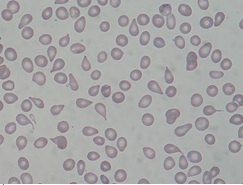
Diagnóstico
El diagnóstico se confirma mediante un estudio de la médula ósea, que presenta un aspirado seco, en virtud de la fibrosis medular y una biopsia que pone de manifiesto la fibrosis tanto reticulínica como colagénica.
Tratamiento
– Trasplante alogénico de precursores hematopoyéticos en personas jóvenes.
– Transfusiones y EPO en casos de anemia grave.
– Andrógenos y folatos que ocasionalmente pueden mejorar la anemia.
– Esplenectomía cuando la esplenomegalia es masiva o produce síntomas graves.
– En fase proliferativa se emplea hidroxiurea y en fase fibrotica se ha utilizado talidomida y esteroides.
Trombocitemia esencial (TE):
Se trata de un síndrome mieloproliferativo crónico, en el que predomina la proliferación megacariocítica-plaquetaria.
En cuanto a la clínica, más de la mitad de los pacientes no presentan síntomas. En el resto de los pacientes se manifiesta fundamentalmente por fenómenos hemorrágicos y/o trombóticos (la manifestación más frecuente de oclusión microvascular es un dolor urente en manos, pies y dedos, denominado eritromelalgia). Puede presentarse esplenomegalia aunque es menos frecuente y excepcionalmente evoluciona a leucemia aguda.
Diagnóstico
El diagnostico de TE se realiza por exclusión. Se basa en una cifra de plaquetas elevada (trombocitosis superior a 500.000 plaquetas por milímetro cúbico), en ausencia de otra causa identificable de trombocitosis (como neoplasias, infección, inflamación crónica, esplenectomía). Para su diagnóstico debemos descartar los demás síndromes mieloproliferativos crónicos (PV, MP y la LMC, todos ellos pueden cursar con trombocitosis), así como la ferropenia (recordar que la ferropenia es causa de trombocitosis secundaria).
Tratamiento
– Pacientes con TE de bajo riesgo (menores de 60 años sin riesgo cardiovascular): no requieren tratamiento.
– Pacientes con TE de alto riesgo: tratamiento mielosupresor (hidroxiurea) hasta alcanzar un recuento plaquetario entre 400.000 y 450.000/dl, AAS en dosis bajas y anagrelide si es necesario.
Leucemia Mieloide Crónica (LMC):
Se trata de una neoplasia mieloproliferativa crónica, en la que predomina la proliferación de la serie mieloide.
Dentro de los síndromes mieloproliferativos crónicos, la LMC es la más frecuente, y aparece habitualmente en personas de 50-60 años, con ligero predominio en el género masculino.
La leucemia mieloide crónica está claramente relacionada con un marcador citogenético, el cromosoma Philadelphia, que aparece hasta en el 95% de los casos. Dicho cromosoma Philadelphia consiste en una translocación del material genético entre los cromosomas 9 y 22, lo que da lugar a la unión del oncogén ABL del cromosoma 9 con el oncogén BCR del cromosoma 22, originando un híbrido anormal BCR/ABL, que es el causante de la enfermedad. Esta región codifica una proteína tirosina-cinasa que facilita la proliferación e inhibe la apoptosis celular, diana terapéutica del mesilato de imatinib.
Entre un 20-40% de los pacientes se encuentran asintomáticos al momento del diagnóstico. En el resto de los pacientes la LMC se puede manifestar como un síndrome hipermetabólico (astenia, perdida de peso y sudoración nocturna), acompañado de hepatoesplenomegalia y un síndrome anémico progresivo.
Diagnostico
Sangre periférica:
– Hemograma: se observa un inceremento de los globulos blancos en todas sus manifestaciones (neutrófilos predominantemente, pero también eosinófilos, basófilos, algunos blastos e incluso monocitos). Respecto a la serie roja, suele existir anemia normo-normo, con un número no elevado de reticulocitos. La serie plaquetaria puede presentar desde trombopenia hasta trombocitosis.
– Bioquímica: es característica la disminución de algunas enzimas de los neutrófilos, tales como la fosfatasa alcalina leucocitaria y la mieloperoxidasa o lactoferrina.
Médula ósea: característicamente hipercelular, con un incremento de la relación mieloide/eritroide.
Citogenética: al diagnóstico, el 95% de los casos de LMC presentan el cromosoma Philadelphia , que puede ser detectado por hibridación in situ por fluorescencia (FISH) o técnicas de PCR.
Fases de la Leucemia mieloide crónica:
Fase crónica: exsite un aumento progresivo de la proliferación de la serie blanca, acompañada de desdiferenciación, o aparición de formas inmaduras en la sangre periférica. Todo ello se acompaña de una anemia progresiva y hepatoesplenomegalia.
Fase acelerada: se observa un incremento llamativo en la hepatoesplenomegalia, al mismo tiempo que se infiltran órganos por las células leucémicas y aumenta el porcentaje de células blásticas, tanto en la sangre periférica como en la médula ósea.
Fase blástica o fase de transformación a leucemia aguda: definida cuando el porcentaje de blastos en la medula ósea es superior al 20% ( 15% en sangre periférica). Durante la fase de transformación, que ocurre en el 80% de los casos, predomina la leucemia aguda de tipo no linfoide o mieloblástica.
Tratamiento
– Solamente es curativo el trasplante alogénico de precursores hematopoyéticos, que presenta mejores resultados en los dos primeros años de enfermedad.
– Si no puede realizarse el trasplante, el tratamiento a elección es el mesilato imatinib, un inhibidor de la proteína tirosina-cinasa BCR/ABL, o el interferón .
Pronóstico
Son factores de peor pronóstico la edad avanzada, un recuento muy elevado de células blancas en la sangre perifrérica, anemia grave, esplenomegalia gigante, alto porcentaje de blastos en medula y sangre, trombocitosis grave y aparición de nuevas alteraciones citogenéticas.
Resumen
- La PV cursa con un aumento de las tres series sanguíneas (con predominio de la serie roja) y esplenomegalia, SIN aumento de la EPO, lo que permite distinguirla de cualquier poliglobulia secundaria.
- El cuadro característico de la MF consiste en anemia progresiva con glóbulos rojos en lágrima o dacriocitos, elementos inmaduros mieloides y eritroides (leucoeritroblastosis) en sangre periférica y esplenomegalia masiva. El diagnóstico se realiza a través de una biopsia de médula ósea.
- La TE suele ser asintomática, aunque puede manifestarse por fenómenos hemorrágicos y/o trombóticos (eritromelalgia) y esplenomegalia moderada. Su diagnóstico se realiza por exclusión.
- La sintomatología típica de la LMC consiste en hepatoesplenomegalia, síndrome anémico progresivo e hipermetabolismo (astenia, pérdida de peso y sudoración nocturna). Además, encontramos leucocitosis, anemia normo-normo, disminución de fosfatasa alcalina leucocitaria y una médula ósea hipercelular.
- La mutación de tirosina-cinasa JAK2 se enceuntra en más del 95% de los casos de PV y en el 50-60% de los casos de TE y MF; mientras que el gen de fusión BCR/ABL, resultado de la t(9,22) en el cromosoma Philadelphia se encuentra en el 95% de los casos de LMC.
- En la fase acelerada de la LMC aumenta la proliferación y las células inmaduras y blastos en sangre, siendo fase blástica cuando cumple criterios de leucemia aguda (20% de blastos en la MO)
- La MF cursa con fibrosis de la MO, provocando un patrón mieloptísico con emigración de las células hematopoyéticas a hígado y bazo.
- La fosfatasa alcalina leucocitaria se encuentra elevada solamente PV, mientras que se encuentra disminuida en LMC y HPN.
- El tratamiento curativo general de los SMPc es el trasplante de médula ósea. Para mielosuprimir (quimioterapia) el tratamiento de elección es la hidroxiurea. Se utiliza interferón en pacientes menores de 50 años y mujeres en edad fértil, ya que la hidroxiurea puede ser dañina para el feto o producir infertilidad.
- El tratamiento específico de elección para cada SMPc es:
– PV: sangrías (flebotomías)
– LMC: mesilato de imatinib
– TE: anagrelida
– MF: esplenectomía
Caso clínico
Hombre de 46 años, sin antecedentes mórbidos, refiere cuadro de 3 meses de evolución caracterizado por compromiso del estado general, baja de peso 8 kg y sudoración nocturna. En el último mes se agrega sensación de plenitud abdominal.
Examen físico revela palidez y Esplenomegalia de 16 cm BRC.
Hemograma:
| Hb (g/dL) | Hematocrito (%) | VCM (fL) | Leucocitos (x103 / mm3) |
Plaquetas (x103 / mm3) |
Reticulocitos (%) | VHS (mm/h) |
| 8 | 24,5 | 82 | 49,8 | 531 | 0,5 | 11 |
| Eosinófilos | Basófilos | Mielocitos | Promielocitos | Juveniles | Baciliformes | Segmentados | Linfocitos | Monocitos | Blastos |
| 1 | 3 | 4 | 3 | 3 | 9 | 72 | 9 | 1 | 1 |
| 1-4 | 0-1 | 0 | 0 | 0 | 0-2 | 60-70 | 20-35 | 4-8 | 0 |
Del perfil bioquímico destaca uricemia de 9 g/dl, LDH 800 U/L y fosfatasa alcalina leucocitaria disminuida.
